Using Patient Feedback to Improve Treatment Outcomes for Patients with Congenital Dyserythropoietic Anaemia Type I Receiving Interferon Therapy
Karl Frey, Sanja Brolih, Caroline Scott, Nicholas Fordham, Sam Burrows, Nyree Cole, Karen Deem, Christopher Jenkins, Melanie Proven, Christian Babbs, Noemi Bernadette Alice Roy

TL;DR
This study improves treatment for a rare blood disorder by incorporating patient feedback on interferon therapy outcomes and side effects.
Contribution
The study introduces a new framework for evaluating interferon therapy in CDA-I that includes patient-reported outcomes.
Findings
IFNα administration significantly increases hemoglobin levels by 30.7 g/L (p < 0.001).
Current studies lack assessment of IFNα's impact on patient symptoms and quality of life.
A new classification framework incorporating patient feedback is proposed for better treatment evaluation.
Abstract
Congenital dyserythropoietic anaemia type-I (CDA-I) is a rare autosomal recessive disease characterised by ineffective erythropoiesis, haemolysis and non-haematological developmental abnormalities. Its treatment is multifactorial, including the management of anaemia, iron overload and prevention of osteoporosis. The only treatment specific to CDA-I is subcutaneous interferon alpha (IFNα) 2A. This study presents the first summary of all published cases of CDA-I patients (n = 33) treated with IFNα and categorises their outcome. We also present new unpublished cases (n = 7). Overall, we find that IFNα administration causes a statistically significant mean increase in haemoglobin of 30.7 g/L (p < 0.001). However, we note that previous studies do not assess the impact of IFNα therapy on providing symptomatic benefit to patients with CDA-I, or the weight of side effects on their quality of…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
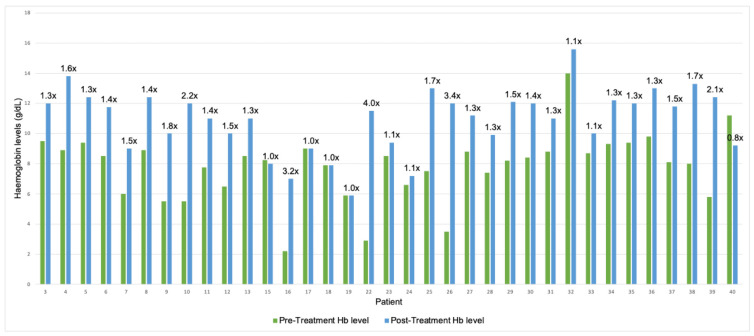 Figure 1
Figure 1- —Congenital Anaemia Network
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsErythrocyte Function and Pathophysiology · Blood disorders and treatments · Blood groups and transfusion
1. Introduction
Congenital dyserythropoietic anaemia type-I (CDA-I) (MIM 607465 and #224120) is a rare autosomal recessive disease characterised by macrocytic anaemia, ineffective erythropoiesis, haemolysis and fairly minor non-haematological developmental abnormalities [1]. Erythroblasts of CDA-I patients display pathognomonic morphological abnormalities that can be identified by electron microscopy (EM), referred to as a ‘spongy’ heterochromatin. Additionally, light microscopy reveals enucleation defects, binuclearity and internuclear bridging. Other non-erythroid haematopoietic lineages are unaffected in CDA-I [2].
While these unique microscopic features appear to be preserved across all patients with CDA-I, there is vast heterogeneity in phenotype expression. A 2006 study by Heimpel et al. showed that the age at diagnosis of 21 patients with CDA-I ranged from 0.1 to 47 years old (median 17.3) [3]. This heterogeneity is not only seen between unrelated patients with different mutations, but even in families with identical mutations. For example, al-Fawaz and al-Mashhadani, 1995, describe two siblings, where one presented symptoms in the neonatal period and the other at 2 years of age [4]. CDA-I patients typically present with symptoms of anaemia, which may be accompanied by jaundice, splenomegaly and complications from iron overload, due to increased iron absorption. Non-haematological abnormalities have also been reported, including syndactyly, short stature, flattened vertebral bodies and early osteoporosis. CDA-I is frequently misdiagnosed as more common congenital anaemias, such as congenital haemolytic anaemia or hereditary spherocytosis [3,5].
At the genetic level, 90% of CDA-I patients harbour biallelic mutations in either of the two known causative genes, CDAN1 and CDIN1 [6]. In the remaining 10% of cases, no mutations can be identified in either of these genes, suggesting the presence of a third disease locus. Roy and Babbs, 2019, report 51 known causative mutations in CDAN1 and 5 in CDIN1 [1]. No genotype-phenotype correlations have been identified thus far. Pathogenic CDAN1 alleles were first discovered among the Israeli Bedouin tribal group, and CDA-I remains most prevalent in this population. Worldwide, the reported incidence of CDA-I suggests a frequency of 0.2–4.8 cases per million live births [7].
The management of CDA-I is multidisciplinary. The anaemia can be treated by regular blood transfusions, and although splenectomy has been used, it is of little benefit [8]. Iron overload is managed with iron chelation therapy, or, in non-anaemic individuals, with regular venesections. Most patients receive folic acid to reverse deficiencies caused by haemolysis. Additional specialist input is required for management of non-haematological features of CDA-I, including syndactyly and osteoporosis [9].
The only available treatment specific to CDA-I is the administration of subcutaneous interferon alpha (IFNα) 2A. It is known to effectively raise haemoglobin (Hb) levels and reduce iron absorption, thereby decreasing or eliminating the need for blood transfusions and iron chelation therapy. This was first found inadvertently in a patient with CDA-I receiving IFNα treatment for hepatitis C [1]. Scott et al., 2022, confirmed administration of IFNα raises Hb levels in CDA-I patients and further demonstrates that CDA-I patients receiving IFNα treatment show an increase in the number of E burst forming units (BFU-Es), which are the first progenitor cells committed to the erythroid lineage [10]. Electron microscopy studies show that treatment with IFNα partially reverses the ‘spongy’ heterochromatin appearance [6]. IFNα is now widely used for CDA-I, but there is no strict evidence base for this or for its dosing regimens, because no clinical trials have been conducted in this patient group and dosing has followed that currently recommended for hepatitis C treatment, with no investigations as to whether an alternative dosing regimen would be better suited in CDA-I. Additionally, IFNα carries a wide side effect profile, which makes it difficult for some patients to tolerate [11].
A recent report published by the James Lind Alliance highlights the Top 10 priorities for Rare Inherited Anaemias as established by a committee of clinicians, researchers, and patients [12]. This includes two points that are highly relevant to this study:
- Would a national formal network of clinicians with expertise and/or a national MDT (multidisciplinary team meeting) improve care for patients with rare inherited anaemias?
- Would a register of all rare inherited anaemia patients in the UK (including data and samples) improve care?
While the National Haemoglobinopathy Registry in England, United Kingdom (UK) should have all CDA-I patients registered, the latest published data purports that there are only 11 patients with CDA-I in England, UK [13], a number that is clearly incorrect and at odds with the predicted 300–400 cases expected from genetic carrier rates [6]. We believe that a formal framework for IFNα therapy in CDA-I would improve patient care and facilitate future research in the field. To achieve this, we collaborate with Congenital Anaemia Network (CAN), an organisation that funds research and supports patients with congenital anaemias to gather the input of people with lived experience of the condition.
2. Methods
In this study, we incorporate a systematic literature review about treatment responses in all CDA-I patients who have been published to have received IFNα. We also include results from unpublished cases. In total, we collected 40 CDA-I patient reports: 33 already published reports, representing our retrospective case series, and 7 unpublished CDA-I patient reports. We perform a paired t-test statistic for Hb levels pre and post treatment with IFNα. Finally, we collaborate directly with CDA-I patients and carers to propose new ways in which the therapeutic assessment in CDA-I could be standardised to improve patient care and facilitate future research. This includes the creation of a formal classification system to record efficacy and side effects of IFNα therapy using patient-reported outcome measures (PROMs).
Published cases: We searched the PubMed database comprising all recent biomedical literature from MEDLINE, life science journals, and online books published in English before 5 April 2021 using the following or a combination of the following terms: “congenital dyserythropoietic anaemia I”; “CDA-I”; “interferon”; “interferon alpha” and/or “interferon alpha2”.
Inclusion/exclusion criteria: Patients included in this analysis were diagnosed by positive genetic tests for known CDA-I genotypes or displayed ‘spongy’ heterochromatin on electron microscopy. Cases with a CDA-I diagnosis based solely on light microscopy were excluded from this study.
Unpublished cases: We present new cases of patients under speciality care at the Cancer and Haematology Centre, Churchill Hospital, Oxford, or those referred to the South-Central Genomic Laboratory Hub for diagnosis. We obtained written consent from patients or those with parental responsibility over patients for the anonymous publication of this data.
Patient involvement: We collaborate directly with six patients or parents of patients with CDA-I undergoing IFNα therapy through the charity ‘Congenital Anaemia Network’ (CAN). We organised two meetings with the aim of encouraging patient representation in the creation of a formal framework for monitoring and recording outcomes to IFNα therapy in CDA-I.
3. Results
In this study, we review 40 patients with CDA-I who have received IFNα therapy and outline their demographics, genetic data, clinical presentation, and response to treatment. The results are outlined in Table 1 [2,3,5,6,13,14,15,16,17,18,19,20,21,22]. A treatment response categorisation criterion is incorporated under the ‘response’ column. This criterion is described in Section 4 and outlined in Table 2.
All patients were treated with variations in IFNα. Dosing regimens varied and did not appear to be protocol-driven. Patients receiving pegylated IFNα typically only required once weekly injections, while non-pegylated IFNα was typically given tri-weekly. We found no trend or correlation between dosage and frequency of IFNα therapy and response, as long as adherence was consistent. In fact, the authors would like to note the vast heterogeneity and at times incomplete patient reports which combined with low patient number demand a cautious interpretation of the data. Nevertheless, from clinical practice we recognise that there is currently poor guidance on how to dose IFNα and in the absence of clinical trial data, this remains empirical and based on doses published for Hepatitis C or myeloproliferative conditions. Pragmatically, many clinicians titrate the dose to optimise response and reduce side effects, but particularly in children, evidence of how to carry this out would be extremely helpful to patients and clinicians alike.
We also found no correlation between the type of mutation and treatment outcomes. Overall, five patients (patients 14, 17, 18, 19, 21) were reported as not having responded to treatment. In the case of patient 14, this can be attributed to low treatment compliance and a self-purchased IFNα. Therefore, this patient was excluded from further data analysis. Patients 17–19 are siblings born to consanguineous parents of Kuwaiti origin, all harbouring the L178Q mutation in CDIN1. Interestingly, patient 16 displays the same mutation but showed a significant increase in Hb upon IFNα, suggesting genetic modifiers may play a role. Patient 21 underwent genetic testing for CDA-I, but no mutations were found in CDAN1 or CDIN1. A diagnosis was made based on electron microscopy. Given that the detection of ‘spongy’ heterochromatin is diagnostic of CDA-I, this finding supports the hypothesis that there is a third disease locus, as suggested by Olijnik et al. 2021 [6]. Intriguingly, this patient did not respond to IFNα, potentially linking poor treatment outcomes to mutations in a yet undiscovered CDA-I disease gene.
Our extensive literature search highlighted that most studies reported outcomes primarily based on pre- and post-treatment Hb levels. Figure 1 highlights the pre- and post-treatment Hb levels where available. In patients where these were documented, we found a mean increase of 30.7 g/L following IFNα therapy, or on average a 1.5-fold increase (mean pre-treatment: 79.0 g/L; mean post-treatment 109.7 g/L; n = 36). We performed a paired t-test to prove statistical significance in this effect (p < 0.001). No other significant conclusions could be drawn from these comparisons, although we note that IFNα treatment at an early age seems to have a more pronounced effect on Hb levels. From our clinical experience, response to IFNα is usually rapid (within 4–8 weeks), and lack of response after this time period usually predicts complete unresponsiveness, and the therapy should probably be stopped at that stage. While most clinicians, especially when starting IFNα in young children who have been transfusion dependent, monitor bloods weekly, there is probably no need to do this, and a monthly blood count should be sufficient. Liver function tests should also be carried out, and a rise in ALT can be seen, which is usually transient but may require dose reduction in IFNα.
Crucially, no study considered patient preferences or a clinically validated PROMs reporting system as useful parameters to define treatment success. We also found that side effect reporting was inconsistent and would benefit from standardisation. We therefore collaborated directly through CAN with six patients (or carers) with CDA-I receiving IFNα therapy. We organised an online discussion forum to collect PROMs and later re-evaluated these at the yearly national CAN CDA-1 patient conference 2025.
Using an online questionnaire, overall experience with IFNα was quantified on a 1–5 Likert scale, with a mean of 3.3/5 (n = 6). Positive effects of IFNα included increased energy levels, reduced need for iron chelation and blood transfusions, and a reduction in vomiting and pain flares. All six patients reported experiencing side effects, namely fever, flu-like symptoms, headaches, muscle pains, diarrhoea, and bruising at the injection site. We asked patients to rank the following four questions relating to their treatment based on patient priority. These were selected according to our previous online discussion with the same patient group. They are outlined below in preferential order:
- Are my symptoms (e.g., fatigue) improving with interferon?
- Can I stop other treatments (e.g., iron chelation, blood transfusions) because of interferon?
- Will interferon reduce my risk of CDA-I-related problems in the future?
- Are my Hb levels better after interferon?
It is noteworthy that the impact of the treatment on Hb levels, the only consistently recorded treatment outcome in publications, is of least importance to patients, suggesting that PROMs should be recorded as standard in future assessments and treatment responses.
4. Discussion
Our survey unanimously highlights that symptomatic improvement is the most important aspect of IFNα therapy for our patients. They described frustration over the heavy emphasis on measuring Hb levels throughout their treatment journey, when this frequently did not correspond to improvements in fatigue. We set out to devise treatment response criteria, partly based on objective quantifiable data highlighted by our literature search and partly based on a subjective assessment of symptoms and side effects, as deemed important to our patient group. This is outlined in Table 2. Table 1 incorporates these scoring criteria under the ‘response’ column.
- Quantifiable criteria
Hb levels provide a quantitative measure of the effect of IFNα. Symptoms of CDA-I can be largely attributed to anaemia, but Hb alone is not sufficient to predict symptoms. Fatigue may result from ineffective haematopoiesis and/or haemolysis in addition to Hb levels. Other blood parameters of interest include iron studies to monitor iron overload. Changes in MCV are unlikely to hold clinical value. A reduction in the requirement for blood transfusions and iron chelation can also be quantified. This was of second highest importance to patients in our survey. Blood transfusions are time-consuming and require frequent intravenous access, and some patients report that IFNα use has given them time back in their daily routine. Additionally, iron chelation therapy has a wide side effect profile, including visual changes, rashes, gastrointestinal upset, dizziness, and abdominal cramping.
- 2.PROMs
Ideally, a clinically validated score should be utilised to quantify fatigue before and after treatment. The FACIT-Fatigue (Functional Assessment of Chronic Illness Therapy-Fatigue) scale assesses self-reported fatigue and its impact on daily activities and function [26]. It was developed for a more precise evaluation of fatigue associated with anaemia in cancer patients, but a recent randomised controlled trial has supported the use of this score in iron deficiency anaemia with good reliability [27]. Further research is required to clinically validate a fatigue score for use in congenital anaemias.
- 3.Side Effects
IFNα is well known to cause a range of side effects, most commonly fever, flu-like symptoms, abdominal pain, and diarrhoea. Only 10/39 patients in this study were reported to tolerate IFNα well, with no dose correlation evident from available data. Our collaboration with the patient group has highlighted that these common IFNα side effects are experienced regularly, with heterogeneity as to whether these attenuate over time. In addition, patients reported they took more precautions ahead of their injections, such as pain relief medication or booked time off work, or in some cases, lowered their dosage to prioritise adherence. In fact, from patient feedback, these side effects pose a real-life obstacle to adherence and should therefore carry significant weight in the response evaluation. Patients also noted that information about side effects in the literature and on patient information leaflets commonly focuses on immediate to short term side effects, while patients receive little information on long term effects of IFNα use. This is likely because IFNα is most commonly used for the treatment of Hepatitis B and C as a 6 to 12-month course. Ideally, both short- and long-term side effects of IFNα therapy would be assessed using a personalised IFNα-therapy side-effects scale. In the absence of such a validated clinical scale, especially for Congenital Anaemias, we suggest that adverse event reporting tools such as the Common Terminology Criteria for Adverse Events (CTCAE) could be used for the categorisation criteria [28].
The use of this categorisation criteria would allow clinicians and researchers to collect data more effectively and better understand how patients respond to IFNα and the reasons behind discontinuation of treatment. It could also be used to evaluate any novel therapies that are studied in CDA-I in the future.
In the absence of PROMs from already published reports, we have used the quantifiable criteria to grade the treatment responses of patients in our study. According to these, 32/39 patients in our data analysis (82%) exhibited an overall positive response to IFNα therapy.
CDA-I remains a rare condition, and the scarcity and heterogeneity of the data demand caution when interpreting results. Still, we believe that the use of our proposed system, which includes PROMS, will promote good practise by providing clinicians with a standardised framework. This will allow qualitative and quantitative evaluation of treatment response, facilitating future research in this field. Moreover, it provides the patient with tangible information about their therapeutic response and empowers them to make autonomous decisions about their care. The clinical validation of a fatigue scale, such as FACIT-fatigue for congenital anaemias, would facilitate PROMs for IFNα therapy in patients with CDA-I. We will collaborate with CAN to achieve this goal.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Roy N.B.A. Babbs C. The pathogenesis, diagnosis and management of congenital dyserythropoietic anaemia type I Br. J. Haematol.201918543644910.1111/bjh.1581730836435 PMC 6519365 · doi ↗ · pubmed ↗
- 2Niss O. Lorsbach R.B. Berger M. Chonat S. Mc Lemore M. Buchbinder D. Mc Cavit T. Shaffer L.G. Simpson J. Schwartz J.H. Congenital dyserythropoietic anemia type I: First report from the Congenital Dyserythropoietic Anemia Registry of North America (CDAR)Blood Cells Mol. Dis.20218710253410.1016/j.bcmd.2020.10253433401150 PMC 8809105 · doi ↗ · pubmed ↗
- 3Heimpel H. Schwarz K. Ebnöther M. Goede J.S. Heydrich D. Kamp T. Plaumann L. Rath B. Roessler J. Schildknecht O. Congenital dyserythropoietic anemia type I (CDA I): Molecular genetics, clinical appearance, and prognosis based on long-term observation Blood 200610733434010.1182/blood-2005-01-042116141353 · doi ↗ · pubmed ↗
- 4Al-Fawaz I.M. Al-Mashhadani S.A. Congenital Dyserythropoietic Anaemia Type I Acta Haematol.199593505310.1159/0002040927725852 · doi ↗ · pubmed ↗
- 5Shalev H. Kapelushnik J. Moser A. Dgany O. Krasnov T. Tamary H. A Comprehensive Study of the Neonatal Manifestations of Congenital Dyserythropoietic Anemia Type IJ. Pediatr. Hematol. Oncol.20042674674810.1097/00043426-200411000-0001115543010 · doi ↗ · pubmed ↗
- 6Olijnik A.-A. A Roy N.B. Scott C. A Marsh J. Brown J. Lauschke K. Ask K. Roberts N. Downes D.J. Brolih S. Genetic and functional insights into CDA-I prevalence and pathogenesis J. Med. Genet.20215818519510.1136/jmedgenet-2020-10688032518175 PMC 7613268 · doi ↗ · pubmed ↗
- 7Iolascon A. Esposito M.R. Russo R. Clinical aspects and pathogenesis of congenital dyserythropoietic anemias: From morphology to molecular approach Haematologica 2012971786179410.3324/haematol.2012.07220723065504 PMC 3590084 · doi ↗ · pubmed ↗
- 8Iolascon A. Andolfo I. Barcellini W. Corcione F. Garçon L. De Franceschi L. Pignata C. Graziadei G. Pospisilova D. Rees D.C. Recommendations regarding splenectomy in hereditary hemolytic anemias Haematologica 20171021304131310.3324/haematol.2016.16116628550188 PMC 5541865 · doi ↗ · pubmed ↗