Role of the Super-Enhancer Component Bromodomain Protein 4 in the Radiation Response of Human Head and Neck Squamous Cell Carcinoma Cells
Nanami Munakata, Hironori Yoshino, Masaharu Hazawa, Eichi Tsuruga

TL;DR
This study explores how the protein BRD4 contributes to radioresistance in head and neck cancer cells and suggests targeting BRD4 could improve cancer treatment outcomes.
Contribution
The study reveals a novel role of BRD4 in maintaining ΔNp63 expression, which contributes to radioresistance in HNSCC cells.
Findings
BRD4 inhibition with ARV-771 or knockdown increases radiosensitivity in HNSCC cells.
BRD4-dependent maintenance of ΔNp63 expression contributes to radioresistance.
BRD4 inhibition reduces cell proliferation and enhances apoptosis in irradiated HNSCC cells.
Abstract
Radiotherapy is an effective treatment for cancer; however, radioresistant cancer cells result in recurrence. Therefore, elucidating the mechanisms of radioresistance is urgently needed. Super-enhancers (SEs) are clusters of enhancers occupied by a high density of master transcription factors, mediators, and bromodomain protein BRD4. Recently, we reported that ΔNp63, an oncogenic transcription factor, promotes radioresistance in human head and neck squamous cell carcinoma (HNSCC) cells. As ΔNp63 establishes SEs in HNSCC cells, SEs may be involved in radioresistance. Here, we investigated the role of the SE component BRD4 in the radiation responses of HNSCC cells using a BRD4 degrader ARV-771 or BRD4 knockdown. First, Western blotting confirmed that ARV-771 decreased BRD4 protein expression. ARV-771 treatment resulted in reduced cell proliferation and enhanced apoptosis in irradiated…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
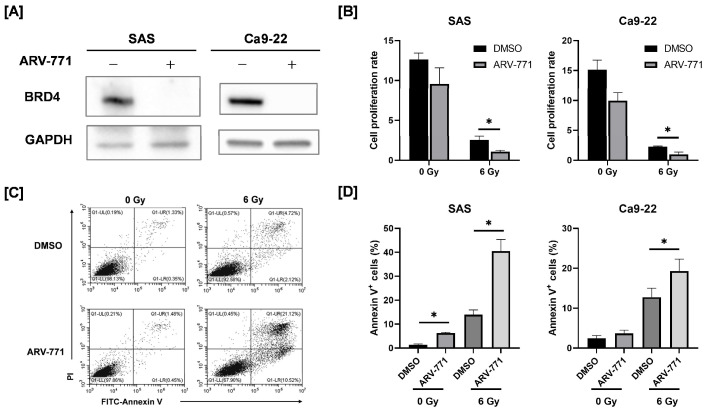 Figure 1
Figure 1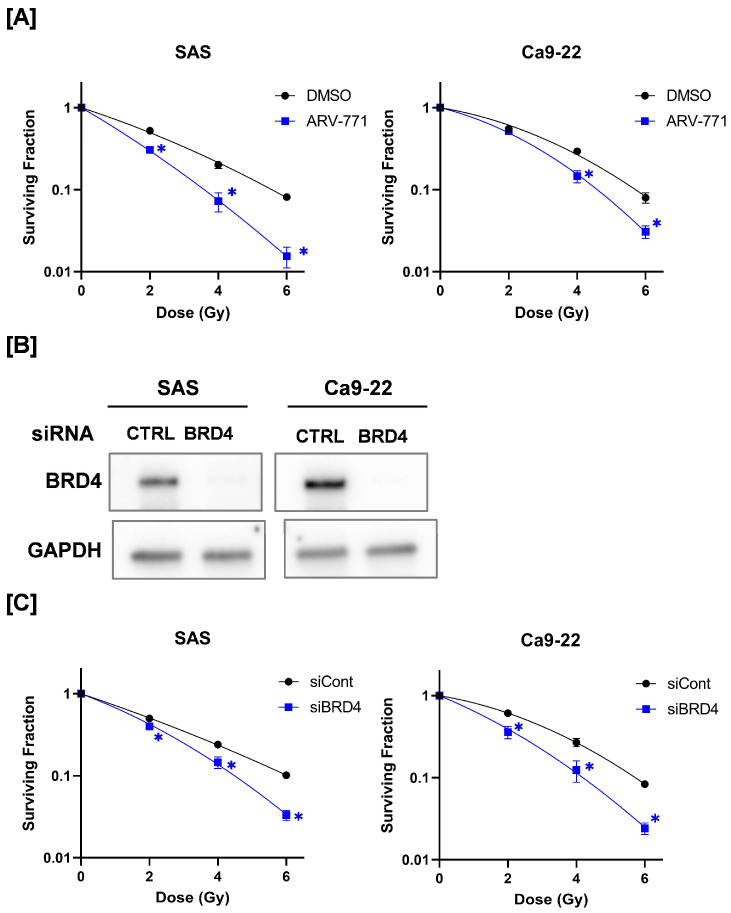 Figure 2
Figure 2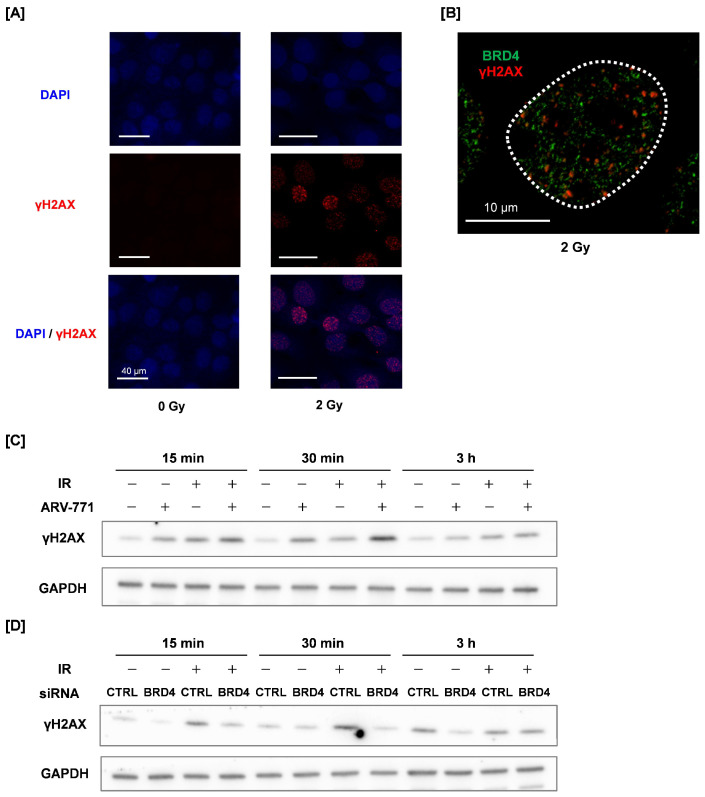 Figure 3
Figure 3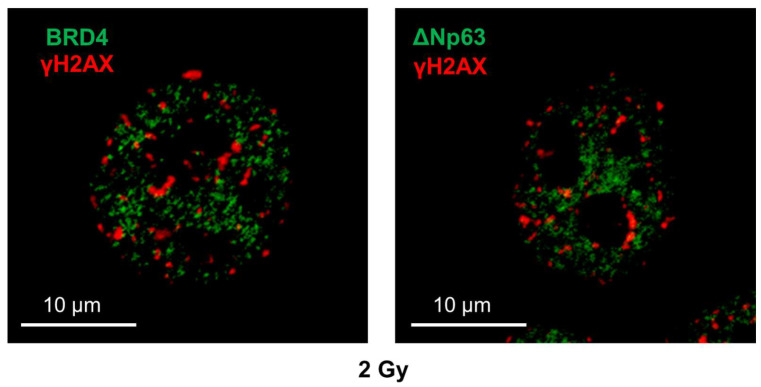 Figure 4
Figure 4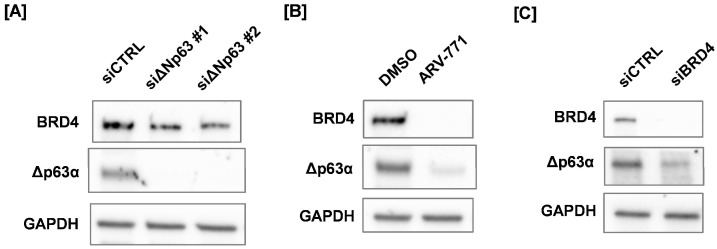 Figure 5
Figure 5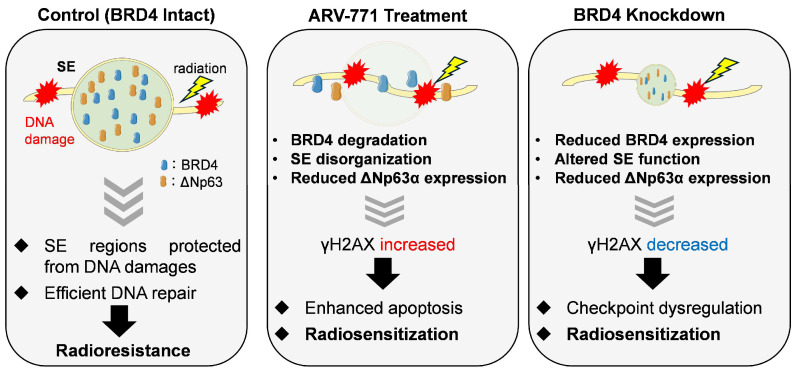 Figure 6
Figure 6- —Mochida Memorial Foundation for Medical and Pharmaceutical Research
- —JSPS KAKENHI
- —Interdisciplinary Collaborative Research Grant for Young Scientists, Hirosaki University
- —Grant for Scientific Research in Master Course in Hirosaki University Graduate School of Health Sciences
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsProtein Degradation and Inhibitors · Chromatin Remodeling and Cancer · Genomics and Chromatin Dynamics
1. Introduction
Most head and neck cancers, collectively known as head and neck squamous cell carcinoma (HNSCC), arise from the mucosal epithelium in the oral cavity, pharynx, and larynx. HNSCC is highly aggressive and heterogeneous [1,2]. Curative treatment with surgery is frequently difficult because the head and neck region contains several vital organs responsible for essential physiological functions, as well as a dense concentration of bones, muscles, nerves, and blood vessels in a relatively small space.
Currently, various approaches are employed for managing HNSCC, including surgery, chemotherapy, radiation therapy, and photodynamic therapy [3]. Among these strategies, radiation therapy is highly effective for localized treatment and is one of the key therapeutic options for HNSCC. However, some cancer cells exhibit radioresistance, resulting in recurrence [4]. Therefore, elucidating the mechanisms of radioresistance and developing strategies to overcome it are crucial for improving prognosis.
We conducted a search for relevant molecular regulators to identify factors involved in radioresistance in HNSCC cells. Consequently, our laboratory recently reported that knockdown of ΔNp63 reduced clonogenic survival of irradiated cells and enhanced radiation-induced apoptosis, indicating a functional role of ΔNp63 in modulating radiation responses [5]. ΔNp63 is an isoform of p63, a member of the p53 tumor suppressor family, and the major isoform of p63 expressed in squamous cell carcinoma (SCC) is ΔNp63α [6]. ΔNp63α serves as an oncogene in SCC and promotes the early steps of SCC development [7]. Kudo et al. reported that ΔNp63α can transcriptionally repress p53 target genes, including BAX and p21, thereby attenuating p53-dependent DNA damage responses following ionizing radiation [8]. Notably, our findings suggest that ΔNp63 contributes to radioresistance even in HNSCC cells harboring mutant p53, suggesting the involvement of p53-independent survival mechanisms [5].
In addition, ΔNp63 is a master transcription factor and a super-enhancer (SE) component. SEs are clusters of enhancers that strongly induce gene expression [9]. They are characterized by histone H3K27 acetylation (H3K27ac), bromodomain protein 4 (BRD4), and the mediator complex [10]. Although typical enhancer regions range from 0.2 to 1 kb in length, SEs are formed when multiple transcription factors bind to the mediator complex at several adjacent enhancers, causing a large enhancer cluster [9]. Although the precise functions of SEs are not yet fully understood, they have gained attention as major regulatory elements impacting cell identity and disease pathogenesis, particularly in cancer [11,12,13]. For example, SE disruption in squamous cell carcinoma cells has been demonstrated to suppress tumorigenicity and metastasis in human HNSCC stem cells [14]. These findings suggest that SEs play a significant role in regulating cancer cell self-renewal and tumorigenesis and can function as potential therapeutic targets in cancer management. Furthermore, since ΔNp63 regulates HNSCC cell radioresistance, SEs may play a role in radioresistance. However, details remain unclear.
BRD4, a member of the bromodomain and extra-terminal domain (BET) protein family, is a key component of SEs. By interacting with acetylated lysine residues in histone proteins, it preferentially binds to active enhancers, contributing to chromatin remodeling and gene transcription [15]. BRD4 mainly promotes RNA polymerase II phosphorylation and mediates RNA polymerase II pausing and elongation [16]. Considering its role in oncogene regulation [17], BRD4 has become a crucial target for cancer therapy, resulting in the development of multiple BET inhibitors [18]. Recently, BET degradation inducers utilizing the ubiquitin–proteasome system, known as proteolysis-targeting chimeras (PROTAC), have been developed and are currently being investigated for clinical applications [19]. Here, we investigated the role of SEs in the radiation response of HNSCC cells using ARV-771 [20,21], a PROTAC BRD4 degrader.
2. Materials and Methods
2.1. Reagents
PBS(−) (Ca^2+^, Mg^2+^-free Dulbecco’s phosphate-buffered saline) and methanol were purchased from Wako Pure Chemical Industries (Osaka, Japan). Propidium iodide (PI), fetal bovine serum (FBS), and dimethyl sulfoxide (DMSO) were purchased from Sigma-Aldrich (St. Louis, MO, USA). ARV-771 was purchased from Med Chem Express (Monmouth Junction, NJ, USA). Anti-BRD4 rabbit antibody (#63759), anti-deltaN p63 (E6Q3O) rabbit antibody (##67825), anti-Glyceraldehyde–3–phosphate dehydrogenase (GAPDH) rabbit antibody (#5174), anti-p-histone H2A.X (γH2AX; #9718), horseradish peroxidase-conjugated anti-rabbit IgG antibody (#7074), horseradish peroxidase-conjugated anti-mouse IgG antibody (#7076), and AlexaFluor 488^®^-conjugated mouse IgG antibody (#4408) were purchased from Cell Signaling Technology Japan, K.K. (Tokyo, Japan). Anti-BRD4 rabbit antibody (Ab128874) was purchased from Abcam plc (Cambridge, UK), anti-γH2AX mouse antibody (JBW301) from Upstate Biotechnology, Inc. (Lake Placid, NY, USA), and CF™ 543-conjugated anti-mouse IgG (H + L) antibody (#20306) from Biotium (Fremont, CA, USA). Anti-human ΔNp63 rabbit antibody (#619001), FITC-Annexin V, and Annexin V binding buffer were purchased from BioLegend (San Diego, CA, USA). Lipofectamine^®^ RNAiMAX, Select Predesigned siRNA against the gene encoding ΔNp63 (#1: no. s16413, #2: no. s16411), BRD4 (no. s22120), and Silencer^®^ Select negative #1 Control siRNA (no. 4390843) were obtained from Thermo Fisher Scientific, Inc. (Waltham, MA, USA).
2.2. Cell Culture and Treatment
Human HNSCC cell lines SAS and Ca9-22 were purchased from RIKEN Bio-Resource Center (Tsukuba, Japan). Cells were cultured in Dulbecco’s Modified Eagle Medium high glucose (Wako) medium containing 10% FBS and 1% penicillin (100 units/mL)–streptomycin (100 μg/mL) (Wako) at 37 °C in a humidified atmosphere containing 5% CO_2_.
Cells were seeded in 35 mm culture dishes at a density of 5.0 × 10^4^ cells/dish and cultured for 2 days. Following incubation, the culture medium was removed, the medium containing DMSO or ARV-771 (final concentration, 1.0 μM) was added, and cells were further cultured for 24 h. The ARV-771–containing medium was removed, and cells were washed with PBS (−). ARV-771 was used at a final concentration of 1.0 μM, which was selected based on previous studies demonstrating efficient BRD4 degradation at this dose [20,21]. Following 1 h incubation, cells were irradiated with X-rays. At 24 h after irradiation, each cell was collected using 0.25% trypsin/Ethylenediaminetetraacetic acid (EDTA) (Wako), counted using the trypan blue dye exclusion method, and the cells were used for each analysis. For proliferative ability evaluation and apoptosis analysis, cells were collected 3 days after replacing cells, counted using the trypan blue dye exclusion method, reseeded onto 35 mm dishes at a density of 5.0 × 10^4^ cells/dish, and cultured for another 3 days.
2.3. SiRNA Transfection
Cells were seeded in 24-well plates at a density of 4.0 × 10^4^ cells and transfected with siRNA targeting ΔNp63 and BRD4 using Lipofectamine^®^ RNAiMAX Transfection Reagent (Invitrogen, Thermo Fisher Scientific, Waltham, MA, USA) according to the manufacturer’s protocol. Following 48 h transfection, cells were harvested for subsequent analysis. The final siRNA concentration was 10 nM.
2.4. In Vitro X-Ray Irradiation
Cells were irradiated using an X-ray generator (MBR-1520R-3; Hitachi, Ltd., Tokyo, Japan) at 450 mm from the focus and at a dose rate of 0.99–1.02 Gy/min (150 kVp; 20 mA; 0.5 mm Al filter and 0.3 mm Cu filter).
2.5. Colony Formation Assay
The irradiated cells were harvested after 24 h of culture and appropriate cell numbers were seeded onto 60 mm culture dishes (Sumitomo Bakelite Co., Ltd., Sumitomo Bakelite Co., Ltd., Tokyo, Japan) Colonies containing more than 50 cells were counted. The surviving fraction was calculated as previously described [22].
2.6. SDS-PAGE and Western Blotting
SDS-PAGE and Western blot analysis were performed as previously described [23]. Equal amounts of protein (about 2 μg per lane) were loaded onto SDS–polyacrylamide gels. The following primary antibodies were used: anti-ΔNp63 antibody (1:3000), anti-BRD4 antibody (1:3000), and anti-GAPDH antibody (1:4000). After overnight dilution at 4 °C, the membrane was reacted with a secondary antibody (1:10,000) for 1 h at 24 °C and detected using chemiluminescence with Clarity Western ECL Substrate (Bio-Rad Laboratories, Inc., Hercules, CA, USA). Images were captured using the iBright 1500 system (Thermo Fisher Scientific, Inc.). GAPDH was used as the loading control.
2.7. Analysis of Apoptosis
Apoptosis was analyzed using FITC-Annexin V and PI staining according to the manufacturer’s instructions. Briefly, cells were harvested, washed twice with PBS (−), centrifuged at 1200 rpm for 5 min at 24 °C, and suspended in 100 μL of Annexin V binding buffer. Five microliters of FITC-Annexin V (90 μg/mL) and PI (1 mg/mL) were added to the cell suspension, and cells were incubated for 15 min at 24 °C in the dark. After adding Annexin V binding buffer, samples were analyzed using a flow cytometer (CytoFLEX; Beckman-Coulter, Inc., Brea, CA, USA).
2.8. Fluorescence Immunostaining
SAS cells were seeded onto 35 mm dishes with submerged coverslips, irradiated with 2 Gy, and incubated for 30 min. Following incubation, cells were fixed in 4% paraformaldehyde (NACALAI TESQUE, INC., Kyoto, Japan) for 15 min at 24 °C. Following fixation, the coverslips were washed with PBS (−), and the samples were blocked with blocking buffer (5% bovine serum albumin/0.3% Triton X-100/PBS [−]) for 1 h. Subsequently, the samples were incubated overnight at 4 °C with the following primary antibody (1:200) in 1% bovine serum albumin/0.3% Triton X-100/PBS (−). Following incubation, the coverslips were washed with PBS (−), and a secondary antibody (1:200) in 1% bovine serum albumin/0.3% Triton X-100/PBS (−) was added and incubated for 1 h at 24 °C. Subsequently, the coverslips were washed with PBS (−), sealed, and mounted on glass slides using Prolong^®^ Gold Antifade Reagent with DAPI (#8961). The samples were analyzed using a confocal laser microscope (LSM 710; Carl Zeiss Co., Ltd., Tokyo, Japan).
2.9. Statistical Analysis
Data were presented as means ± standard error of three independent experiments. Comparison of the two groups was performed using Student’s t-test. A p-value of <0.05 was considered statistically significant. Statistical analysis was performed using Excel (Microsoft 365, Washington, DC, USA) with the add-in software Statcel4 (The Publishing OMS Ltd., Tokyo, Japan).
3. Results
3.1. Proliferation and Apoptosis in Non- or X-Ray–Irradiated HNSCC Cells Treated with ARV-771
We treated HNSCC cells with the BRD4 degrader ARV-771 and analyzed BRD4 protein expression to investigate the role of BRD4 in the cellular radiation response of HNSCC. ARV-771-treated cells exhibited lower BRD4 expression than DMSO-treated controls (Figure 1A).
Subsequently, we investigated cell proliferation and apoptosis in ARV-771–treated HNSCC cells under non- and X-ray–irradiated conditions. ARV-771 showed minimal impact on the proliferation rate of non-irradiated HNSCC cells (Figure 1B). However, 6 Gy–irradiated cells demonstrated significantly reduced proliferation at days 3 and 6 for SAS and Ca9-22 cells, respectively (Figure 1B). Moreover, apoptosis analysis revealed that ARV-771–treated cells had a higher percentage of apoptotic cells following 6 Gy irradiation than DMSO controls at the corresponding time points (Figure 1C,D).
3.2. Effects of ARV-771 or BRD4 Knockdown on Radiosensitivity of HNSCC Cells
We evaluated the radiosensitivity of ARV-771–treated HNSCC cells using a colony formation assay. As shown in Figure 2A, the surviving fractions of SAS and Ca9-22 cells were lower in the ARV-771-treated group than those in the DMSO-treated group, suggesting that ARV-771 enhances HNSCC cell radiosensitivity (Figure 2A). Similarly, BRD4 knockdown enhanced the radiosensitivity of HNSCC cells (Figure 2B,C). Taken together, these results indicate that BRD4 is involved in the radioresistance of HNSCC cells.
To evaluate whether ARV-771 or BRD4 knockdown affects radiosensitivity through alterations in cell cycle distribution, we performed cell cycle analysis following ARV-771 treatment and BRD4 knockdown. As shown in Supplementary Figure S1A, ARV-771 treatment increased the G2/M population in SAS cells, whereas it increased the G1 population in Ca9-22 cells. In contrast, BRD4 knockdown consistently increased the G1 population in both SAS and Ca9-22 cells (Supplementary Figure S1B). Importantly, an increase in the G1-phase population is generally associated with relative radioresistance compared with G2/M-phase cells [24]. Despite this, both ARV-771 treatment and BRD4 knockdown enhanced radiosensitivity in our experiments (Figure 2). These findings suggest that the observed changes in radiosensitivity cannot be sufficiently explained by alterations in cell cycle distribution alone.
3.3. Association of BRD4 with DNA Double-Strand Breaks (DSBs) in X-Ray–Irradiated SAS Cells
As DNA damage triggers radiation-induced cellular responses [22], we subsequently analyzed the association between DNA damage and BRD4. We investigated the localization of γH2AX and BRD4. γH2AX is widely used as a sensitive marker of DSBs; however, it also plays an essential role in the DNA damage response by serving as a platform for the recruitment and amplification of DNA repair and checkpoint signaling factors, including MDC1, 53BP1, and BRCA1 [25,26].
As shown in Figure 3A, γH2AX foci are observed in SAS cells at 30 min following 2 Gy irradiation, whereas they are rarely detected in non-irradiated cells. Notably, confocal laser microscopy analysis revealed that the signal distribution of γH2AX and BRD4 in 2 Gy–irradiated SAS cells was mutually exclusive (Figure 3B). This finding suggests that DSBs do not occur in the SE region. Therefore, it is suggested that the SE region is a DNA damage-resistant genomic region.
We evaluated the γH2AX levels by Western blotting when ARV-771 was combined with irradiation. ARV-771 treatment markedly enhanced radiation-induced γH2AX levels compared with irradiation alone (Figure 3C), indicating increased DNA damage accumulation when BRD4-mediated SE function was disrupted. These results suggest the notion that SE-associated chromatin states contribute to limiting DNA damage accumulation following irradiation. In addition, ARV-771 treatment alone increased basal γH2AX levels even in the absence of irradiation (Figure 3C), suggesting that BRD4 degradation induces endogenous DNA damage or enhances DNA damage signaling. Upon irradiation, ARV-771 further augmented γH2AX accumulation, consistent with an increased DNA damage burden and/or impaired resolution of DNA lesions. In contrast, BRD4 knockdown attenuated radiation-induced γH2AX induction (Figure 3D). Notably, the opposite effects of ARV-771 treatment and BRD4 knockdown on γH2AX accumulation suggest that pharmacological BRD4 degradation and genetic BRD4 depletion influence DNA damage responses through distinct mechanisms. These divergent effects indicate that γH2AX levels reflect not only DNA repair kinetics but also BRD4-dependent chromatin context and DNA damage signaling capacity.
3.4. Association Between BRD4 and ΔNp63 in Irradiated SAS Cells
We previously reported that the knockdown of ΔNp63, an SE component, improved HNSCC cell radiosensitivity [5]. Therefore, we analyzed the association between BRD4 and ΔNp63 in the irradiated SAS cells. Initially, we analyzed γH2AX and ΔNp63 localization. ΔNp63 and γH2AX, as well as BRD4 and γH2AX, exhibited different subnuclear localization in 2 Gy–irradiated SAS cells (Figure 4). This finding suggests that DSBs cannot occur in the regions where ΔNp63 or BRD4 is highly expressed in the irradiated cells.
Next, we analyzed BRD4 and ΔNp63α protein expression in ΔNp63 knockdown or ARV-771-treated SAS cells. ΔNp63 knockdown effectively reduced ΔNp63α protein expression but had little effect on BRD4 protein levels (Figure 5A). In contrast, ARV-771 treatment reduced both BRD4 and ΔNp63α protein expression (Figure 5B). In addition, BRD4 knockdown resulted in efficient depletion of BRD4 and was accompanied by a reduction in ΔNp63α protein levels (Figure 5C). Taken together, these results suggest that BRD4 contributes to the maintenance of ΔNp63α expression.
4. Discussion
Elucidating the mechanisms of radioresistance and their regulation in cancer cells is essential for improving the efficacy of radiotherapy. In this study, we focused on BRD4, a key SE component, to elucidate the role of SEs in HNSCC cell radioresistance. Our findings showed that ARV-771–treated HNSCC cells with reduced BRD4 protein expression exhibited high radiosensitivity and low proliferation following irradiation. In addition, BRD4 knockdown enhanced radiosensitivity of HNSCC cells. These results suggest the role of BRD4 in HNSCC cell radioresistance. Furthermore, we noted that γH2AX foci were not observed at BRD4-enriched sites in irradiated cells, suggesting that radiation-induced DSBs do not occur in SE regions where BRD4 is highly expressed. Collectively, BRD4 may regulate HNSCC cell radioresistance by protecting the genomic regions involved in the survival from radiation.
Notably, γH2AX levels were differentially affected by pharmacological BRD4 degradation and BRD4 knockdown. ARV-771 increased basal γH2AX levels even in the absence of irradiation and further enhanced γH2AX accumulation after irradiation, whereas BRD4 knockdown attenuated radiation-induced γH2AX induction. These contrasting effects suggest that γH2AX reflects not only the extent of DNA double-strand breaks but also BRD4-dependent chromatin context and DNA damage signaling capacity. In this regard, BRD4 degradation by ARV-771 may induce endogenous DNA damage and/or potentiate DNA damage signaling by disrupting transcription-associated chromatin dynamics. Consistent with this interpretation, BRD4 has been shown to suppress R-loop accumulation and transcription–replication conflicts, and its loss results in increased DNA damage and γH2AX accumulation even in the absence of exogenous genotoxic stress [27]. In contrast, BRD4 knockdown may reduce γH2AX accumulation through distinct mechanisms, potentially involving transcriptional reprogramming of DNA damage response factors or adaptive changes in chromatin organization. Indeed, BRD4 regulates multiple aspects of genome maintenance beyond transcriptional control, including replication stress responses and chromatin-mediated DNA damage signaling pathways [17]. Thus, the divergent effects of BRD4 degradation and knockdown on γH2AX levels likely reflect fundamental differences in how BRD4 loss is achieved and how chromatin-associated DNA damage responses are engaged. At present, it remains unclear whether these effects represent direct modulation of DNA damage induction or indirect consequences of altered DNA damage response pathways, and additional analyses using complementary DNA damage indicators will be required to fully elucidate the mechanisms by which BRD4 regulates radiation responses.
Accumulating epigenomic evidence has revealed extensive reprogramming of SE landscapes in SCC, including HNSCC, leading to aberrant activation of oncogenic transcriptional programs that regulate tumor cell identity, stemness, and therapeutic resistance [28,29]. BRD4 has been shown to preferentially occupy SE regions in SCC models and to drive the expression of key oncogenes and lineage-defining transcription factors, such as TP63/ΔNp63 and MET [21,30]. Genome-wide enhancer profiling further demonstrates that p63 preferentially binds SEs and cooperates with chromatin regulators including BRD4 to sustain oncogenic transcriptional states in SCC cells [31,32]. SE-associated transcriptional programs have been implicated in aggressive tumor phenotypes, metastasis, and resistance to anticancer therapies, and BRD4 is frequently overexpressed in HNSCC, correlating with poor clinical outcomes [33]. Although direct clinical evidence linking SE enrichment or BRD4 genomic occupancy to radioresistance in HNSCC patients remains limited, these epigenomic observations collectively support a mechanistic framework in which BRD4-dependent SE function contributes to cellular responses to therapeutic stress, including ionizing radiation. Our findings extend this framework by providing functional evidence that disruption of BRD4-dependent SE activity enhances radiation-induced DNA damage responses in HNSCC cells.
Beyond chromatin-associated mechanisms, physical properties of SE-associated condensates may also influence DNA damage susceptibility. Recent evidence has indicated that liquid–liquid phase separation (LLPS) is involved in SE formation [34,35]. LLPS is a biological and physical phenomenon wherein a homogeneous liquid phase separates into two distinct liquid phases, similar to how oil and water separate. In biological systems, LLPS is involved in organizing cellular components without the necessity for membrane-bound organelles [36] and regulating cellular functions, including transcription [37]. We reported that the interior of LLPS condensates is a diluted aqueous environment [38]. Since low linear energy transfer (LET) radiation, including X-rays, induces DNA damage primarily through radicals generated by the radiolysis of water molecules, accounting for 60–70% of the damage [39], it is possible that X-rays cannot induce DSBs in SE regions formed by LLPS due to the lower water content. In contrast, high-LET radiation, including α-rays, can induce DNA damage independently of radical formation. Therefore, evaluating whether high-LET radiation can induce DNA damage in SE regions is necessary.
Similarly to BRD4, ΔNp63 localization was excluded from γH2AX-positive regions in irradiated cells, suggesting that genomic regions occupied by ΔNp63 may be relatively protected from radiation-induced DNA damage. We observed that ARV-771 treatment reduced ΔNp63α protein expression, whereas ΔNp63 knockdown had little effect on BRD4 protein levels. In addition, BRD4 knockdown was accompanied by a reduction in ΔNp63α expression. Consistent with these observations, we previously reported that ΔNp63 knockdown enhances radiation-induced apoptosis and radiosensitivity in HNSCC cells [5], which are also observed following ARV-771 treatment. Taken together, these findings suggest that BRD4-dependent maintenance of ΔNp63α expression may contribute, at least in part, to the regulation of radiation-induced apoptosis and radioresistance in HNSCC cells, although additional studies will be required to determine whether this relationship is direct or mediated through intermediate factors.
To integrate these observations, we propose a schematic model illustrating the differential effects of pharmacological BRD4 degradation and genetic BRD4 knockdown on SE integrity and DNA damage responses (Figure 6). Under basal conditions, BRD4 cooperates with ΔNp63 at SEs to sustain transcriptional programs involved in cell survival and DNA repair, thereby contributing to radioresistance. ARV-771–mediated BRD4 degradation induces rapid SE collapse, leading to transcriptional stress and aberrant DNA damage signaling, manifested as elevated γH2AX levels even in the absence of irradiation. In contrast, BRD4 knockdown partially preserves SE structures but disrupts the proper initiation and spatial organization of DNA damage response signaling, resulting in attenuated γH2AX induction following irradiation despite increased radiosensitivity. Thus, γH2AX reflects distinct underlying processes depending on the mode of BRD4 inhibition, highlighting the context-dependent role of BRD4 in coordinating SE architecture and DNA damage responses. This model is further supported by previous studies demonstrating a functional linkage between BRD4 and ΔNp63α in stratified squamous epithelial cells. Foffi et al. reported that BRD4 expression is positively correlated with p63 expression in human keratinocytes [40], supporting a role for BRD4 as an SE-associated co-activator of lineage-defining transcription factors. In this context, ARV-771–mediated BRD4 degradation may lead to reduced ΔNp63 expression by disrupting BRD4-dependent transcriptional programs.
The relevance of BRD4-dependent SE regulation may extend beyond HNSCC. Recently, we reported that ARV-771 enhances the radiosensitivity of human lung cancer cell line A549 [41]. Notably, A549 cells do not express ΔNp63 [42], suggesting that ARV-771 enhances A549 cell radiosensitivity independently of ΔNp63. In lung cancer cells, transcription factors, such as SMAD3, have been reported to constitute SEs [43,44]. Therefore, other SE-related transcription factors may be involved in radioresistance in lung cancer cells. As the transcription factors constituting SEs are different among cancer types, identifying those involved in radioresistance may result in reduced radioresistance. Notably, however, ARV-771 has been demonstrated to overcome radioresistance in some cancer types, thereby suggesting that targeting BRD4 can effectively regulate gene expression regardless of the specific transcription factor involved.
In conclusion, the results of this study suggest that the SE component BRD4 plays an important role in regulating radioresistance in HNSCC cells, potentially by protecting ge-nomic regions critical for cell survival from radiation-induced damage. Although the pre-cise molecular mechanisms linking BRD4-dependent SE function to DNA damage induction and repair remain to be fully clarified, our findings provide evidence that modulation of BRD4 alters cellular radiation responses. Further studies aimed at dissecting the mechanistic basis of SE-mediated radioresistance, incorporating additional DNA damage and repair assays as well as clinically relevant models, will be essential for establishing the therapeutic potential of targeting BRD4 to improve radiotherapy.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Chen S.M.Y. Krinsky A.L. Woolaver R.A. Wang X. Chen Z. Wang J.H. Tumor immune microenvironment in head and neck cancers Mol. Carcinog.20205976677410.1002/mc.2316232017286 PMC 7282929 · doi ↗ · pubmed ↗
- 2Johnson D.E. Burtness B. Leemans C.R. Lui V.W.Y. Bauman J.E. Grandis J.R. Head and neck squamous cell carcinoma Nat. Rev. Dis. Primers 202069210.1038/s 41572-020-00224-333243986 PMC 7944998 · doi ↗ · pubmed ↗
- 3Jiang Z. Yang X. Ainiwaer M. Chen F. Liu J. Recent clinical and preclinical advances in external stimuli-responsive therapies for head and neck squamous cell carcinoma J. Clin. Med.20221217310.3390/jcm 1201017336614974 PMC 9821160 · doi ↗ · pubmed ↗
- 4Zhang H. Wang X. Ma Y. Zhang Q. Liu R. Luo H. Wang Z. Review of possible mechanisms of radiotherapy resistance in cervical cancer Front. Oncol.202313116498510.3389/fonc.2023.116498537692844 PMC 10484717 · doi ↗ · pubmed ↗
- 5Sato K. Yoshino H. Sato Y. Nakano M. Tsuruga E. ΔNp 63 regulates radioresistance in human head and neck squamous carcinoma cells Curr. Issues Mol. Biol.2023456262627110.3390/cimb 4508039437623213 PMC 10453785 · doi ↗ · pubmed ↗
- 6Cancer Genome Atlas Network Comprehensive genomic characterization of head and neck squamous cell carcinomas Nature 201551757658210.1038/nature 1412925631445 PMC 4311405 · doi ↗ · pubmed ↗
- 7Devos M. Gilbert B. Denecker G. Leurs K. Mc Guire C. Lemeire K. Hochepied T. Vuylsteke M. Lambert J. Van Den Broecke C. Elevated ΔNp 63α levels facilitate epidermal and biliary oncogenic transformation J. Investig. Dermatol.201713749450510.1016/j.jid.2016.09.02627725202 · doi ↗ · pubmed ↗
- 8Kudo K.I. Tsuyama N. Nagata K. Imaoka T. Iizuka D. Sugai-Takahashi M. Muramatsu M. Sakai A. ΔNp 63α transcriptionally represses p 53 target genes involved in the radiation-induced DNA damage response: ΔNp 63α may cause genomic instability in epithelial stem cells Radiat. Oncol.20221718310.1186/s 13014-022-02139-736380314 PMC 9667649 · doi ↗ · pubmed ↗