Plasmablasts as Translational Biomarkers in Autoimmune Diseases: From Cellular Dynamics to Clinical Decision-Making
Muhammad Soyfoo, Julie Sarrand

TL;DR
Plasmablasts are short-lived immune cells that reflect real-time immune activity and could help guide treatment decisions in autoimmune diseases.
Contribution
The paper introduces plasmablasts as dynamic biomarkers for precision immune surveillance in autoimmune diseases.
Findings
Plasmablast kinetics after B-cell-depleting therapies predict relapse in multiple autoimmune conditions.
Single-cell technologies reveal new plasmablast subsets and metabolic states with prognostic value.
Plasmablasts integrate antigenic stimulation and cytokine-driven differentiation into a measurable clinical signal.
Abstract
B cells are key drivers of immune dysregulation across systemic autoimmune diseases. Among their progeny, plasmablasts occupy a uniquely revealing niche: short-lived, highly proliferative intermediates that mirror real-time B-cell activation. Their appearance in peripheral blood integrates antigenic stimulation, cytokine-driven differentiation, and aberrant germinal-center dynamics, transforming them into sensitive indicators of ongoing immunological activity. This review synthesizes current knowledge on plasmablast biology and highlights disease-specific phenotypes across systemic lupus erythematosus (SLE), primary Sjögren disease (pSjD), IgG4-related disease (IgG4-RD), ANCA-associated vasculitis (AAV), and rheumatoid arthritis (RA). We incorporate molecular insights from single-cell technologies that have uncovered previously unrecognized plasmablast subsets, metabolic states, and…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
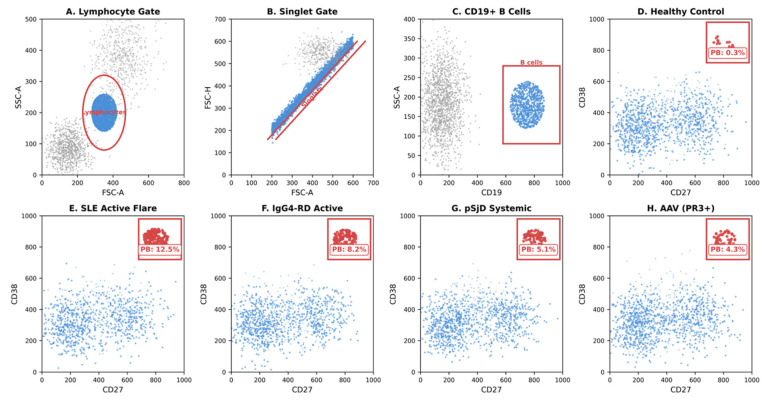 Figure 1
Figure 1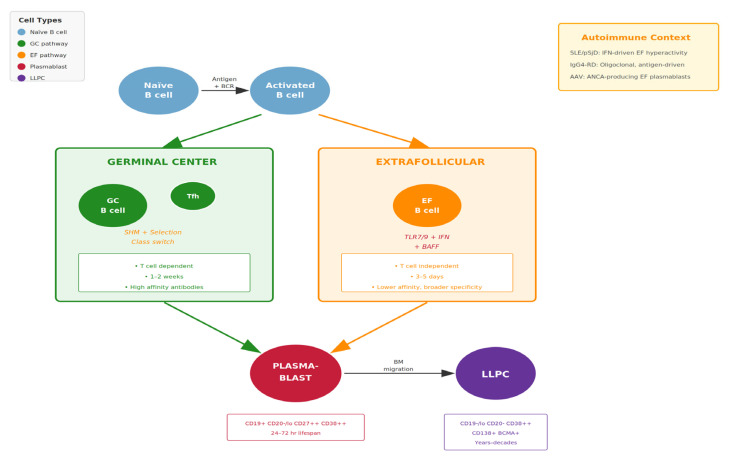 Figure 2
Figure 2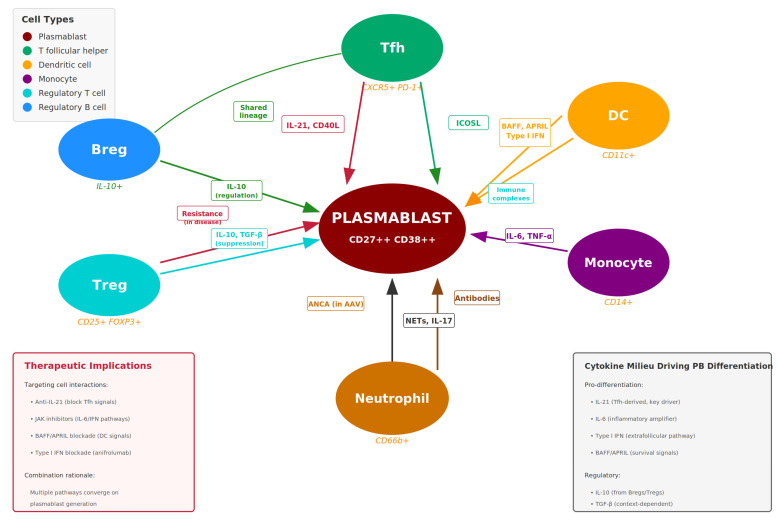 Figure 3
Figure 3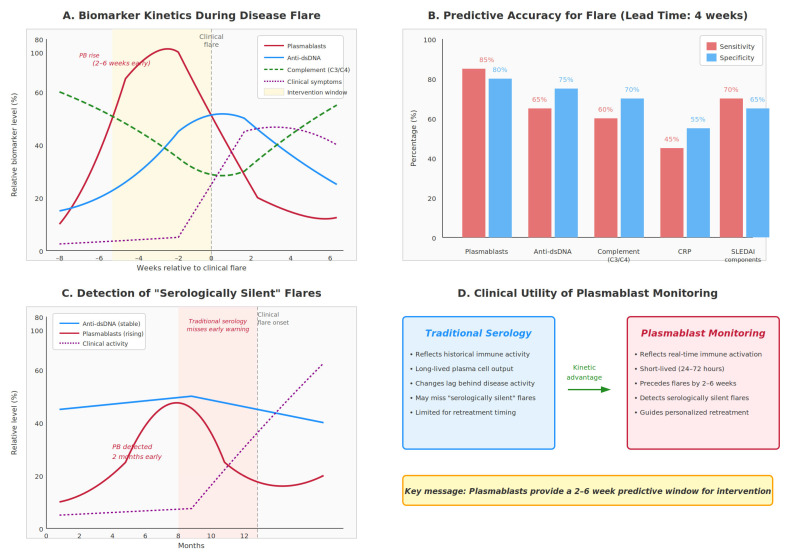 Figure 4
Figure 4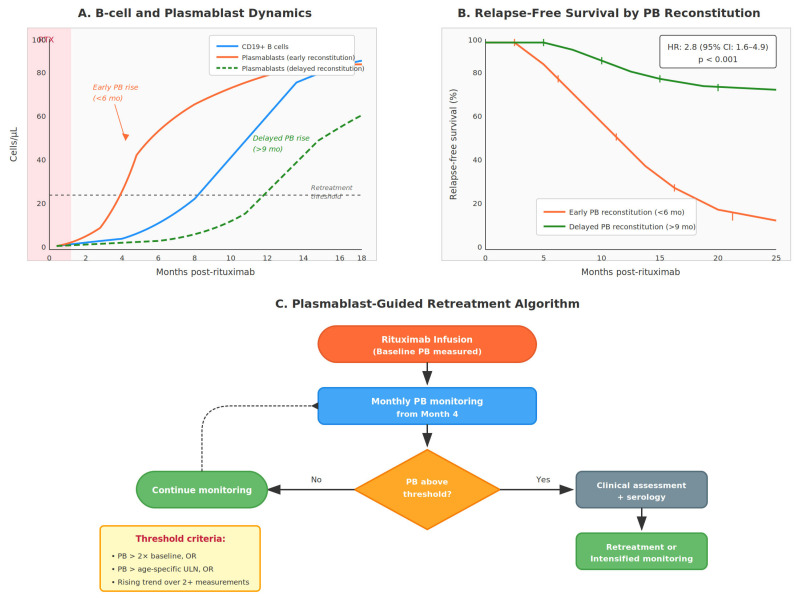 Figure 5
Figure 5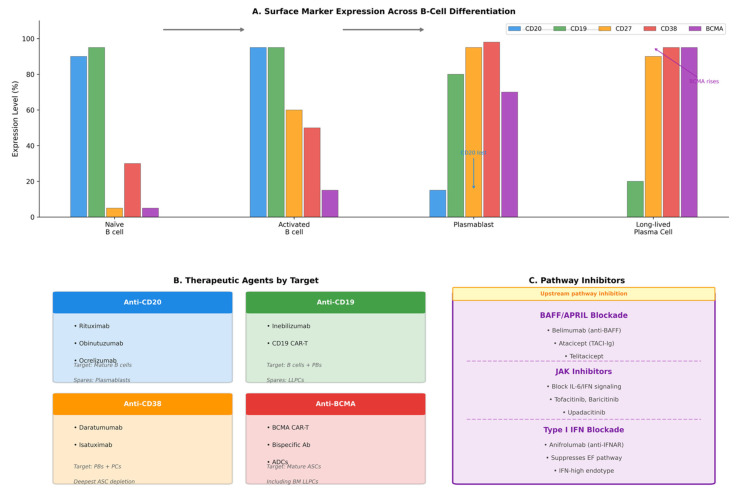 Figure 6
Figure 6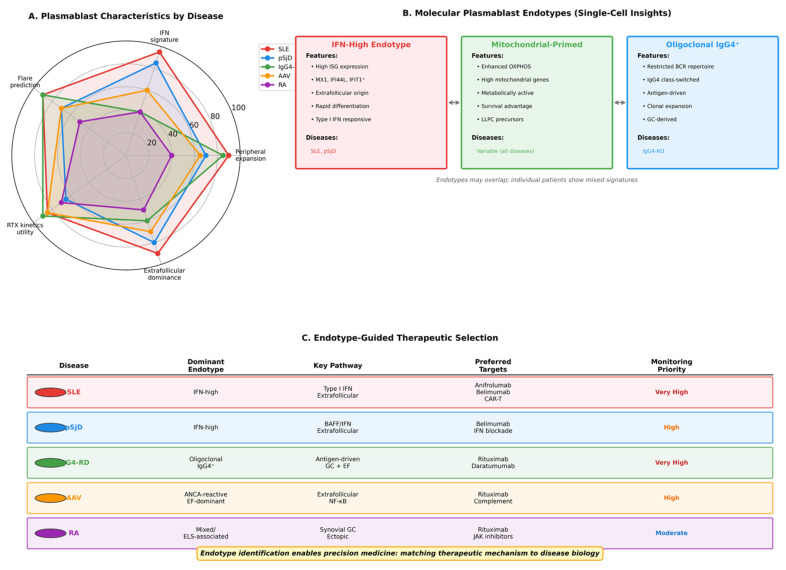 Figure 7
Figure 7Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsSystemic Lupus Erythematosus Research · IgG4-Related and Inflammatory Diseases · Diabetes and associated disorders
1. Introduction
B cells have progressively gained prominence as central orchestrators of immune dysregulation across systemic autoimmune diseases [1,2,3]. Once viewed primarily as antibody factories, B cells are now recognized as key drivers of antigen presentation, cytokine production, and the dynamic propagation of chronic inflammation [4,5]. The success of B-cell-depleting therapies such as rituximab across multiple autoimmune conditions has underscored the pathogenic relevance of the B-cell compartment and stimulated intense interest in understanding the nuances of B-cell biology in disease [6,7].
Within this complex network, plasmablasts occupy a uniquely revealing position. As short-lived, highly proliferative progeny of activated B cells, plasmablasts provide a real-time biological readout of ongoing immune activation [8,9,10]. Their emergence in peripheral blood reflects active differentiation pathways that integrate antigenic stimulation, cytokine signaling—particularly type I interferons and interleukin-21 (IL-21)—and dysregulated germinal center reactions [11,12]. Unlike long-lived plasma cells that reside in protective bone marrow niches and maintain stable antibody titers over decades [13,14], plasmablasts represent the dynamic, transient arm of humoral immunity [15,16]. Importantly, plasmablasts do not exist in isolation but participate in complex bidirectional interactions with other immune cell subsets, including T follicular helper cells, dendritic cells, monocytes, and regulatory T cells, which together shape the magnitude and quality of humoral responses [4,5].
Over the last decade, advances in flow cytometry, mass cytometry (CyTOF), and single-cell multi-omics technologies have redefined plasmablasts as biomarkers of exceptional translational value [17,18]. Across systemic lupus erythematosus (SLE), Sjögren disease (SjD), IgG4-related disease (IgG4-RD), ANCA-associated vasculitis (AAV), and subsets of rheumatoid arthritis (RA), plasmablast expansion correlates with disease activity, flare onset, and therapeutic response [19,20,21]. In particular, the kinetics of plasmablast reconstitution after B-cell-depleting therapy such as rituximab has emerged as one of the most powerful predictors of relapse, often surpassing traditional serological markers in predictive accuracy [22,23,24,25].
The clinical implications of these observations are profound. Whereas autoantibody titers represent a historical record of immune activation—reflecting the cumulative output of long-lived plasma cells—plasmablast levels provide a window into current immunological activity. This kinetic advantage translates into earlier detection of impending flares, more precise timing of retreatment, and the potential for preventive rather than reactive therapeutic interventions. The emergence of therapies specifically targeting plasmablasts and plasma cells, including anti-CD38 monoclonal antibodies and BCMA-directed agents [26,27], further elevates the clinical relevance of understanding plasmablast biology.
This review examines the biology of plasmablasts, their disease-specific phenotypes, and their expanding role as dynamic biomarkers guiding real-time clinical decisions. We discuss how integrating plasmablasts into routine care could shift immune monitoring from static serology to kinetic precision immunology, and we propose practical algorithms for clinical implementation.
2. Biological Overview of Plasmablasts
2.1. Definition and Phenotypic Markers
Plasmablasts occupy a transitional stage between activated B cells and long-lived plasma cells [1,2]. They are typically defined by expression of CD19+CD27+CD38+ high, loss of surface IgD, and increasing expression of transcription factors IRF4, Blimp-1 (encoded by PRDM1), and XBP1 [3,28]. Their proliferation index is among the highest in the immune system, with a half-life of only 24–72 h in circulation [8]. This brevity distinguishes them fundamentally from long-lived plasma cells, which can persist for decades in bone marrow niches [13,14].
The surface phenotype of plasmablasts exhibits both conserved and context-dependent features [10,12]. Core markers include high CD38 expression (typically 10-fold higher than activated B cells), bright CD27 positivity, and variable CD19 expression that diminishes as cells mature toward plasma cell identity. CD20 expression is typically low or absent, explaining why plasmablasts are relatively spared during initial rituximab therapy but are affected by CD19-directed agents [29,30]. Additional markers of clinical relevance include CD138 (syndecan-1), which increases with maturation; HLA-DR, which remains high on plasmablasts but decreases on mature plasma cells; and Ki-67, reflecting their proliferative state [31,32,33]. A standardized flow cytometry gating strategy is illustrated in Figure 1.
Unlike long-lived plasma cells residing in bone marrow niches, plasmablasts remain migratory and responsive to immunological cues [15]. They express chemokine receptors including CXCR4 and CXCR3 that direct their trafficking to inflammatory sites and bone marrow. Their abundance fluctuates rapidly in response to antigen exposure, cytokine stimulation, and local inflammation, making them exquisitely sensitive indicators of immune perturbation [16,17,18].
2.2. Differentiation Pathways
Plasmablasts arise via two principal pathways with distinct immunological characteristics and clinical implications [5,34,35]:
1/Germinal center (GC) pathway: This canonical pathway is induced by T follicular helper cells (Tfh), IL-21, CD40 ligand (CD40L), and sustained antigen presentation [5,36]. Within germinal centers, B cells undergo iterative cycles of somatic hypermutation and affinity-based selection, yielding high-affinity, class-switched immunoglobulins. Plasmablasts emerging from this pathway typically produce antibodies of superior quality and contribute to protective immunity. The process requires approximately 1–2 weeks and generates both plasmablasts destined for short-term antibody production and precursors of long-lived plasma cells [2,37].
2/Extrafollicular pathway: This rapid differentiation route bypasses germinal center reactions and is driven by Toll-like receptor (TLR) activation, B-cell activating factor (BAFF), a proliferation-inducing ligand (APRIL), and interferon-rich environments [35,38,39]. Plasmablasts can emerge within 3–5 days via this pathway, enabling rapid antibody responses to pathogens. However, the extrafollicular pathway is also prominently active in autoimmune diseases, particularly SLE, SjD, and during viral infections [40,41]. Antibodies produced via this pathway undergo less affinity maturation and may exhibit broader—and potentially autoreactive—specificities.
The balance between these pathways has significant implications for autoimmunity. In SLE, the extrafollicular pathway is hyperactive, driven by chronic type I interferon exposure and TLR7/9 stimulation by nucleic acid-containing immune complexes [35,42,43,44]. This results in continuous generation of plasmablasts producing pathogenic autoantibodies. Understanding which pathway predominates in individual patients may guide therapeutic selection, as different agents target distinct aspects of B-cell activation and differentiation [40,45,46] (Figure 2).
2.3. Plasmablast Interactions with Other Immune Cell Subsets
Plasmablasts do not function in isolation but engage in complex, bidirectional crosstalk with multiple immune cell populations that profoundly influence their generation, survival, and effector functions [4,5,46]. Understanding these interactions is essential for interpreting plasmablast dynamics in disease and predicting responses to immunotherapy (Figure 3).
T follicular helper (Tfh) cells provide critical support for plasmablast differentiation through IL-21 secretion, CD40L engagement, and ICOS-ICOSL interactions [35,53]. Aberrant expansion of circulating Tfh-like cells correlates with plasmablast levels across multiple autoimmune diseases, suggesting coordinated dysregulation of the T-B cell axis. In SLE, the Tfh–plasmablast interaction is amplified by the interferon-rich milieu, creating a feed-forward loop of B-cell activation [40,41].
Dendritic cells (DCs) and monocytes contribute to plasmablast generation through multiple mechanisms. Type I interferon-producing plasmacytoid DCs (pDCs) are particularly relevant in SLE and pSjD, where their chronic activation sustains the interferon signature that drives extrafollicular plasmablast differentiation [41,42]. Myeloid DCs present autoantigens and provide co-stimulatory signals, while monocyte-derived cells in inflammatory tissues can support local plasmablast survival through BAFF and APRIL production [54].
Regulatory T cells (Tregs) and regulatory B cells (Bregs) normally restrain plasmablast responses, and their dysfunction contributes to autoimmune B-cell hyperactivity [4,55]. Effective immunotherapy may restore regulatory circuits alongside depleting pathogenic plasmablasts, contributing to durable remissions observed with some treatments.
Neutrophils and other innate immune cells interact with plasmablasts through neutrophil extracellular traps (NETs), which expose autoantigens and activate TLRs on B cells. In AAV, this neutrophil–B cell interaction contributes to both ANCA production and disease pathogenesis [56,57].
These cellular interactions are significantly altered by immunotherapies. B-cell depletion with rituximab disrupts T-B cell collaboration and reduces plasmablast generation, while JAK inhibitors interfere with cytokine signaling networks that support plasmablast differentiation [58,59]. Understanding how different therapies reshape the immune cell landscape helps explain variable responses and informs combination treatment strategies.
3. Plasmablasts Across Autoimmune Diseases
The following sections summarize disease-specific plasmablast associations. It is important to note that the evidence base varies considerably across diseases. Studies in SLE generally comprise larger, multicenter cohorts with prospective designs, while data in IgG4-RD and AAV often derive from smaller, single-center cross-sectional studies. Where possible, we indicate the strength of evidence supporting each association and should therefore be interpreted in the context of study limitations.
3.1. Systemic Lupus Erythematosus
SLE is the best-characterized setting for plasmablast biology, with extensive literature documenting their pathogenic relevance and biomarker utility [19,50,53]. The central role of B cells in SLE pathogenesis is evidenced by the presence of pathogenic autoantibodies, the efficacy of B-cell depletion therapy, and the correlation between B-cell abnormalities and disease activity [49,60]. Multiple studies, including large multicenter cohorts, have demonstrated robust correlations between plasmablast levels and disease parameters [17,19,61]:
Plasmablast surges precede clinical flares by 2–6 weeks, providing a valuable predictive window for intervention [19,62]. Moreover, plasmablast levels correlate with SLEDAI scores, anti-dsDNA antibody titers, complement consumption (low C3/C4), and active nephritis [53,61,63]. These dynamics are driven by an interferon-dependent extrafollicular B-cell activation cascade, linking the type I interferon signature to sustained plasmablastogenesis and B-cell hyperactivity [42,43,44]. Persistent plasmablast elevation identifies patients with refractory disease and a higher risk of cumulative organ damage [64,65]. Therapeutically, the kinetics of B-cell reconstitution following rituximab—particularly early plasmablast reappearance—predict shorter remission duration, with relapse occurring 3–9 months earlier in patients whose plasmablasts return within 4–6 months after treatment [66,67,68]. In contrast, prolonged plasmablast suppression is associated with sustained disease control and reduced glucocorticoid requirements [69,70]. More recently, the therapeutic landscape has expanded substantially, with anti-CD38 therapy (daratumumab) demonstrating notable efficacy in refractory SLE [26] and CD19-directed CAR-T cell therapy achieving unprecedented drug-free remissions in multiply refractory patients [27,71,72]. In lupus nephritis, plasmablast levels correlate with renal activity and proteinuria, and their persistence during treatment predicts incomplete renal response [60,63]. Single-cell and tissue-based studies have further demonstrated plasmablast infiltration within affected kidneys, supporting a direct contribution to local antibody production and immune-mediated tissue damage [49,60].
3.2. Sjögren Disease
Sjögren disease (SjD) displays a distinctive plasmablast signature reflecting its unique pathophysiology characterized by glandular lymphocytic infiltration, B-cell hyperactivity, and increased lymphoma risk [47,73]. The disease is marked by chronic stimulation of the B-cell compartment, elevated serum immunoglobulins, and frequent autoantibody production including anti-SSA/Ro and anti-SSB/La [48,74]. In Sjögren’s disease (SjD), plasmablast biology displays distinct but overlapping features with systemic lupus erythematosus. Several studies have shown that plasmablast expansion correlates primarily with systemic disease activity, as measured by the ESSDAI, rather than with sicca symptoms alone [73,75,76]. Elevated plasmablast levels reflect sustained extrafollicular B-cell activation in a BAFF-rich milieu, with circulating BAFF concentrations correlating closely with plasmablast numbers [77,78]. Importantly, persistent plasmablast elevation has been associated with an increased risk of lymphoma development in selected patient subsets, particularly those exhibiting chronic B-cell activation and germinal center–like structures within salivary glands [79,80,81]. At the transcriptional level, circulating plasmablasts in SjD display a pronounced type I interferon signature, mirroring pathways observed in SLE and reinforcing the concept of shared interferon-driven extrafollicular circuits across systemic autoimmune diseases [47,73]. Notably, plasmablasts frequently remain detectable or rapidly reconstitute following rituximab therapy, likely reflecting the persistence of extrafollicular activation foci within salivary glands that are relatively shielded from circulating anti-CD20 antibodies [82,83,84,85,86]. This phenomenon suggests that peripheral blood plasmablast monitoring may underestimate residual tissue-level disease activity and underscores the need for multicompartment assessment strategies in SjD.
Importantly, plasmablasts may persist or rapidly reappear following rituximab therapy in SjD, reflecting ongoing extrafollicular activation and tissue-resident immune niches rather than incomplete B-cell depletion. This observation highlights a key limitation of plasmablasts as universal therapy-response biomarkers. In this context, persistent plasmablast detection should be interpreted as evidence of compartmentalized immune activity rather than treatment failure per se. These findings underscore the context-dependent nature of plasmablasts as biomarkers and the need for disease-specific interpretation frameworks [83,84,85,86,87].
3.3. IgG4-Related Disease
In IgG4-RD, plasmablasts represent perhaps the most diagnostically and prognostically informative biomarker available [55,87,88,89]. Unlike other autoimmune conditions where plasmablasts serve as one of several useful markers, in IgG4-RD they occupy a central position in disease assessment and monitoring.
In IgG4-related disease (IgG4-RD), plasmablasts exhibit a set of highly distinctive features that confer exceptional diagnostic and disease-monitoring value. Circulating plasmablast levels are often markedly elevated even when serum IgG4 concentrations remain within the normal range, providing superior sensitivity for disease detection compared with serum IgG4 alone [55,90,91]. Plasmablast frequencies correlate strongly with disease activity as assessed by the IgG4-RD Responder Index, frequently exceeding the performance of serum IgG4 itself as a biomarker (based on prospective cohort studies, though sample sizes remain modest) [55,90]. Clonal analyses have demonstrated oligoclonal plasmablast expansions with restricted immunoglobulin heavy-chain variable region usage, supporting an antigen-driven pathogenic process [56,92]. Therapeutically, plasmablast counts decline rapidly following glucocorticoid or rituximab treatment, and early normalization is predictive of sustained clinical remission [93,94]. The particular utility of plasmablast monitoring in IgG4-RD reflects the well-recognized limitations of serum IgG4, which is normal in up to 30% of histologically confirmed cases and may be elevated in a range of unrelated inflammatory or neoplastic conditions [88,95]. In this context, plasmablast enumeration provides critical complementary diagnostic information and can help resolve clinically ambiguous presentations [55,91,96].
3.4. ANCA-Associated Vasculitis
Plasmablasts contribute significantly to the immunopathology of ANCA-associated vasculitis, particularly granulomatosis with polyangiitis (GPA) and microscopic polyangiitis (MPA) [57,58]. The pathogenic role of ANCA—autoantibodies directed against proteinase 3 (PR3) or myeloperoxidase (MPO)—in neutrophil activation and endothelial damage provides a direct link between B-cell–derived antibodies and disease manifestations [58,59].
In ANCA-associated vasculitis (AAV), plasmablast involvement is particularly prominent in PR3-positive disease, where circulating plasmablast levels correlate closely with ANCA titers and markers of immunological activity (based primarily on single-center studies requiring validation) [59,97]. Plasmablast-derived antibodies have been linked to relapse propensity, and persistently elevated plasmablast counts during clinical remission identify patients at increased risk of subsequent disease relapse [97,98]. Multi-omics profiling has revealed enhanced antigen-presentation programs and NF-κB pathway activation in vasculitis-associated plasmablasts, supporting their role as active immunopathogenic effectors rather than passive antibody producers [59]. From a therapeutic perspective, longitudinal monitoring of plasmablast kinetics following rituximab therapy may enable more individualized retreatment strategies in AAV [99,100,101,102,103]. This approach is particularly relevant in granulomatosis with polyangiitis (GPA), where relapse rates remain substantial despite maintenance therapy. In this context, the MAINRITSAN trials have established rituximab as an effective agent for remission maintenance, and integration of plasmablast monitoring may further refine dosing schedules to optimize long-term disease control while minimizing cumulative immunosuppression [102,103].
3.5. Rheumatoid Arthritis
RA shows more modest peripheral blood plasmablast elevations compared to SLE, reflecting a disease process primarily centered within synovial tissue rather than diffuse systemic B-cell activation [104,105]. Nevertheless, plasmablasts play a pivotal role in RA, particularly in seropositive disease and in the context of B-cell–depleting therapy [104,106]. Transient plasmablast peaks coincide with synovial disease exacerbation in seropositive RA, correlating with joint inflammation and structural progression [102,106].
Within inflamed synovium, plasmablasts participate in ectopic lymphoid structures (ELS), where local germinal center–like reactions support autoantibody production and perpetuate inflammation [51,52,107]. The rapid reappearance of plasmablasts following B-cell depletion predicts early clinical relapse, mirroring patterns observed in SLE [69,70].
Moreover, anti-citrullinated protein antibody (ACPA) titers correlate with plasmablast-derived antibody production, underscoring their contribution to pathogenic humoral immunity in RA [108,109].ELS formation, present in approximately 40% of patients with RA, creates specialized microenvironments containing follicular dendritic cells, T follicular helper cells, and actively differentiating B cells, thereby sustaining local plasmablast differentiation and tissue-restricted autoimmunity [107,110].
Table 1 summarizes the comparative characteristics of plasmablasts across these five autoimmune diseases, highlighting differences in peripheral expansion, interferon signature, predominant differentiation pathway, disease activity correlation, flare prediction utility, and response to rituximab kinetics monitoring.
4. Plasmablasts as Real-Time Biomarkers
4.1. Advantages over Traditional Serology
Plasmablasts reflect current immune activation, whereas autoantibody titers represent a historical record produced substantially by long-lived plasma cells [13,14,15]. This fundamental distinction underlies the unique biomarker value of plasmablasts.
Because plasmablasts have exceptionally short lifespans, estimated at approximately 24–72 h, fluctuations in their peripheral blood frequency precede multiple downstream immunological and clinical events [8,17,18]. In several systemic autoimmune diseases, plasmablast surges anticipate overt clinical flares by approximately 2–6 weeks, providing an actionable early warning of impending disease exacerbation [19,62]. These early cellular changes are followed by serologic shifts, including rising autoantibody titers, reflecting the time required for plasmablast-derived antibodies to accumulate in the circulation [17]. Subsequent immune complex formation drives complement consumption, typically manifested by declining C3 and C4 levels [6]. Finally, inflammatory cascades culminate in elevation of acute-phase reactants such as C-reactive protein and erythrocyte sedimentation rate, although this response may be attenuated in interferon-dominant disease states [42].
However, several important caveats must be considered when interpreting plasmablast elevations. Transient plasmablast expansion occurs physiologically following acute infections, vaccinations, and non-disease-related immune activation [18,111]. A recent viral infection can increase plasmablast counts for 1–2 weeks, while vaccination (particularly with mRNA vaccines) may elevate plasmablasts for up to 4 weeks [111]. These confounders can generate false-positive signals if not recognized. Clinicians should ascertain vaccination history and exclude intercurrent infections before attributing plasmablast rises to autoimmune disease activity. Serial measurements demonstrating sustained elevation, rather than single time-point assessments, provide greater specificity for identifying disease-related plasmablast expansion. Additionally, some medications and chronic infections can alter baseline plasmablast levels, necessitating individualized interpretation based on patient context.
This kinetic advantage positions plasmablasts as one of the earliest indicators of loss of disease control. Traditional monitoring strategies relying on quarterly autoantibody titers may miss evolving flares until tissue damage has already occurred [31,32]. Plasmablast monitoring could enable earlier intervention, potentially reducing cumulative organ damage.
4.2. Predicting Flare
Numerous studies have identified plasmablast expansion as one of the strongest predictors of disease flare in systemic lupus erythematosus and IgG4-related disease [19,55,90]. In SLE, increases in circulating plasmablasts precede clinical flares more consistently than conventional biomarkers such as anti–double-stranded DNA antibodies, complement consumption, or overt clinical symptoms [61,62]. The reproducible predictive window of approximately 2–6 weeks observed across multiple cohorts provides a critical opportunity for anticipatory, rather than reactive, therapeutic intervention.
Notably, plasmablast monitoring remains informative even during so-called “serologically silent” flares, in which traditional laboratory markers remain stable despite clinical worsening [19]. In these situations, long-lived plasma cells may sustain relatively stable baseline autoantibody titers, whereas newly generated short-lived plasmablasts produce pathogenic antibodies that drive inflammation before significantly altering total antibody levels, thereby explaining the superior sensitivity of plasmablast dynamics in capturing imminent disease activity [17,64] (Figure 4).
4.3. Monitoring Response to Therapy
Different immunomodulatory agents influence plasmablast biology through distinct and partially complementary mechanisms, and longitudinal tracking of plasmablast dynamics can therefore provide valuable mechanistic insight into therapeutic response [66,67,68,111]. Rituximab, a CD20-directed monoclonal antibody, effectively depletes mature B cells while initially sparing CD20 low/negative plasmablasts; notably, delayed plasmablast reconstitution beyond 6–9 months following treatment is associated with prolonged clinical remission [66,67,68]. Belimumab, by neutralizing soluble BAFF, preferentially attenuates extrafollicular plasmablastogenesis while relatively preserving germinal center–dependent immune responses [112,113,114]. Janus kinase inhibitors dampen cytokine-driven differentiation pathways, particularly IL-21- and T follicular helper cell-dependent signals that are critical for plasmablast generation [115]. Type I interferon pathway blockade, exemplified by anifrolumab, suppresses interferon-high plasmablast subsets that are especially prominent in systemic lupus erythematosus [45,46]. In contrast to these upstream modulatory approaches, anti-CD38 therapy with daratumumab directly depletes plasmablasts and plasma cells via antibody-dependent cellular cytotoxicity, resulting in rapid suppression of antibody-secreting compartments [26]. These differences underscore that plasmablast kinetics must be interpreted in the context of the therapeutic mechanism of action, rather than as a uniform marker of treatment response.
Beyond their role as biomarkers, plasmablasts actively participate in the cytokine networks that sustain and amplify autoimmune inflammation. Activated plasmablasts secrete not only immunoglobulins but also pro-inflammatory cytokines, including interleukin-6 (IL-6), tumor necrosis factor-α (TNF-α), and lymphotoxin, thereby directly contributing to local and systemic inflammatory circuits [4,54]. Conversely, plasmablast differentiation and survival are exquisitely sensitive to the surrounding cytokine milieu. Key signals such as IL-21, IL-6, type I interferons, B-cell activating factor (BAFF), and a proliferation-inducing ligand (APRIL) collectively promote extrafollicular B-cell activation and plasmablast persistence, establishing self-reinforcing inflammatory loops [34,37,39].
This bidirectional cytokine–plasmablast interplay has important implications for both disease flares and therapeutic complications. Massive plasmablast activation, as observed during severe autoimmune exacerbations or following certain immunomodulatory interventions, may contribute to cytokine release syndrome (CRS)–like phenomena characterized by elevated IL-6, interferon-γ, and other inflammatory mediators [116,117]. Although classical CRS is most frequently associated with CAR-T cell therapy, attenuated cytokine release can accompany vigorous plasmablast responses in autoimmune settings. Awareness of this mechanism may help clinicians anticipate and manage inflammatory complications. Conversely, therapies targeting cytokine pathways—such as IL-6 blockade, Janus kinase inhibition, or interferon receptor antagonism—may exert part of their therapeutic benefit through indirect modulation of plasmablast differentiation, survival, and effector function [44,45,59].
4.4. Personalizing Retreatment Schedules
The conventional fixed 6-month rituximab retreatment schedule widely used across autoimmune diseases is largely empirical, originating from early clinical trial designs rather than from immunological principles [99,100]. Marked inter-individual variability exists in both the depth and duration of B-cell depletion and in the kinetics of immune reconstitution following therapy [66,116]. Plasmablast-guided retreatment strategies offer a means to address this heterogeneity by avoiding unnecessary early retreatment in patients with sustained B-cell suppression, thereby reducing cumulative immunosuppression, while simultaneously identifying individuals at higher risk of early relapse who may benefit from shortened retreatment intervals [69,70,90,98,102,103]. By incorporating individualized plasmablast thresholds and reconstitution kinetics, this approach shifts B-cell depletion from a rigid, time-based paradigm toward a dynamic precision-medicine strategy tailored to each patient’s immunological trajectory (Figure 5). The feasibility of such tailored dosing has been demonstrated in ANCA-associated vasculitis by the MAINRITSAN2 trial, providing clinical proof-of-concept for biomarker-guided rituximab retreatment [102].
5. Therapeutic Targeting of Plasmablasts
5.1. CD19-Directed Therapies
CD19 is expressed across the B-cell lineage from early pro-B cells through plasmablasts, but expression diminishes on long-lived plasma cells [29]. This expression pattern makes CD19 an attractive target for depleting the plasmablast compartment while relatively preserving established humoral memory.
Inebilizumab, a humanized monoclonal antibody targeting CD19, induces broad depletion across the B-cell lineage and has been shown to achieve more profound suppression of circulating plasmablast compartments than rituximab, which spares CD19^+^ plasmablasts and plasma cell precursors [30,117,118]. Its clinical efficacy has been most convincingly demonstrated in neuromyelitis optica spectrum disorder (NMOSD), where it is FDA-approved following the N-MOmentum trial, which showed a 77% reduction in attack risk compared with placebo (p < 0.0001) [29,119]. Beyond NMOSD, CD19-directed therapy is being actively explored in several antibody-mediated autoimmune diseases, including systemic lupus erythematosus, ANCA-associated vasculitis, IgG4-related disease, and myasthenia gravis [117]. Targeting CD19 may be particularly advantageous in clinical settings where rituximab proves insufficiently effective, such as in patients with persistent plasmablast populations despite apparent depletion of CD20^+^ B cells, highlighting the relevance of plasmablast biology for therapeutic stratification [29,117].
5.2. CD38-Directed Depletion
CD38 is highly expressed on plasmablasts and plasma cells, making it an attractive target for depleting antibody-secreting cells. Daratumumab, an anti-CD38 monoclonal antibody initially developed for multiple myeloma, demonstrated notable activity across several refractory autoimmune conditions [26]. Although early reports of anti-CD38 therapy in refractory autoimmune disease are encouraging, available evidence is limited to case series and small observational cohorts. Long-term efficacy, infection risk, and durability of immune reconstitution remain incompletely defined. Consequently, CD38-directed therapy should currently be considered experimental and reserved for highly selected, refractory cases.
In systemic lupus erythematosus, case series and small prospective studies have reported dramatic clinical and serological responses in patients who had failed multiple conventional and biologic therapies (total n < 50 patients reported) [26]. However, randomized controlled trial data are currently lacking, and longer-term safety in this population remains to be established
Similar benefit has been described in relapsing or refractory ANCA-associated vasculitis, supporting a role for plasma cell–directed therapy in severe disease (case reports and small series) [120]. In IgG4-related disease characterized by plasma cell–rich tissue infiltrates, daratumumab has been associated with deep and durable responses, further highlighting the pathogenic relevance of antibody-secreting cells in this condition (Limited case reports) [91]. However, depletion of both plasmablasts and long-lived plasma cells carries specific risks, most notably hypogammaglobulinemia and increased susceptibility to infections, necessitating careful monitoring and, in selected cases, immunoglobulin replacement therapy [26,119,121]. The optimal dosing, duration, and patient selection criteria for daratumumab in autoimmune diseases require definition through prospective clinical trials currently in development.
5.3. BCMA-Targeted Therapies
B-cell maturation antigen (BCMA, also known as TNFRSF17) is selectively expressed on plasmablasts and plasma cells and plays a critical role in their survival through interactions with BAFF and APRIL [122,123,124]. This restricted expression profile has positioned BCMA as an especially attractive therapeutic target for eliminating autoantibody-producing cells. Several BCMA-directed strategies are currently being explored, including chimeric antigen receptor (CAR) T-cell therapies, bispecific T-cell engagers that redirect cytotoxic T cells via simultaneous binding to BCMA and CD3, and antibody–drug conjugates delivering cytotoxic payloads selectively to antibody-secreting cells [27,72]. Among these approaches, BCMA-directed CAR-T cell therapy has generated particular enthusiasm in systemic lupus erythematosus, where early reports in patients with severe, multiply refractory disease (n < 30 patients in published series) have described profound immunological reset and clinical responses, including apparent drug-free remissions following a single infusion, with durability extending beyond 12 months in some cases [27,71,72]. These preliminary data, while encouraging, derive from single-center case series without randomized comparators and require validation in larger controlled trials.
However, several important considerations temper immediate extrapolation to clinical practice: (1) published data represent highly selected patients from specialized centers; (2) long-term durability beyond 2 years remains unknown; (3) manufacturing requirements, costs, and access barriers limit current applicability; (4) cytokine release syndrome and immune effector cell-associated neurotoxicity syndrome (ICANS), though generally manageable, require intensive monitoring; and (5) infectious complications from prolonged hypogammaglobulinemia require further characterization.
The broader landscape of CAR-T cell target antigens continues to evolve rapidly. Beyond CD19 and BCMA, numerous cancer-specific antigens are under investigation, and insights from hematologic malignancy treatment inform potential applications in autoimmunity [125]. The selection of optimal target antigens—balancing efficacy against autoreactive cells with preservation of protective immunity—remains an active area of research that will shape the future of cellular therapy in autoimmune diseases.
In conclusion, BCMA-targeted approaches, including CAR-T cells, represent a promising but still early therapeutic strategy in autoimmunity. While dramatic responses have been reported in isolated cases, experience remains limited and extrapolation from oncology warrants caution. Antigen specificity, off-target effects, and long-term immune consequences require careful evaluation, as highlighted in recent analyses of cancer-specific target antigens [125].
5.4. Combined Strategies and Future Directions
Future therapeutic regimens in systemic autoimmune diseases are likely to rely on rational combinations of agents targeting distinct and complementary aspects of B-cell biology in order to achieve synergistic and more durable disease control [112,113,114,115,125,126]. One promising strategy involves combining B-cell depletion approaches, such as rituximab or anti-CD19 therapies, with blockade of the type I interferon pathway to simultaneously suppress autoreactive B-cell populations and the upstream inflammatory signals that sustain their activation [45,46,127]. Another approach pairs anti-CD38–mediated depletion of plasmablasts and plasma cells with BAFF inhibition, aiming both to eliminate existing antibody-secreting cells and to prevent their regeneration from precursor pools [112,113,114]. Finally, BCMA-directed therapies may be integrated with antigen-specific tolerization strategies to achieve deep, targeted elimination of pathogenic plasma cells while promoting long-term immune reprogramming and sustained immunological tolerance [27,67,68]. Several clinical trials are actively evaluating plasmablast- and plasma cell-targeted therapies in autoimmune diseases. Notable examples include: (1) randomized trials of daratumumab in SLE and systemic sclerosis; (2) phase 2 studies of BCMA-targeted CAR-T cells in lupus nephritis; (3) trials combining rituximab with belimumab (BLISS-BELIEVE, NCT03312907); and (4) studies of next-generation anti-CD19 agents in various autoimmune conditions. These trials will provide crucial data on efficacy, safety, optimal patient selection, and the role of plasmablast monitoring in guiding therapy.
These approaches aim to recalibrate the entire B-cell axis, achieving not merely suppression of autoimmunity but potentially durable tolerance induction [68,115] (Figure 6).
6. Integrating Plasmablasts into Clinical Algorithms
6.1. Standardization Challenges
Before plasmablast monitoring can be incorporated into clinical guidelines, several methodological challenges require consensus and standardization across centers [31,32,33,119,121]. A major issue relates to gating strategies, as consistent identification of plasmablasts remains difficult due to progressive CD19 downregulation on mature subsets, necessitating harmonized flow cytometry panels and analytical frameworks [31,33]. Marker selection further influences assay performance, with different combinations of CD27 and CD38, and optional inclusion of CD138, BCMA, or Ki-67, affecting sensitivity, specificity, and biological interpretation [32,119]. In addition, lack of standardized reference intervals and the confounding effects of age, sex, ethnicity, and baseline immune status, are major challenges to multicenter comparisons [31,121]. Several confounding factors, including acute infections, recent vaccinations, and certain immunomodulatory therapies, can induce transient plasmablast expansions unrelated to autoimmune disease activity [18,128]. These limitations currently restrict the widespread clinical implementation of plasmablast monitoring and emphasize the need for multicenter harmonization initiatives. Encouragingly, large multicenter harmonization efforts are underway, and international initiatives such as the EuroFlow Consortium and the Human Immunology Project have developed standardized immunophenotyping panels that could be readily adapted for robust and reproducible plasmablast monitoring in clinical practice [32,33].
6.2. Practical Algorithm for Clinicians
A pragmatic clinical workflow for integrating plasmablast monitoring into autoimmune disease management can be envisioned across several key stages. At baseline, either at diagnosis or treatment initiation, plasmablast levels should be assessed alongside conventional serological markers, including autoantibodies, complement fractions, immunoglobulin levels, and, when available, interferon activity scores, in order to establish an individual reference profile [31,42,53]. During clinical evaluation of suspected disease flare, repeat plasmablast measurement may provide supportive evidence of active immunological disease when levels rise above the patient’s baseline [19,55,62]. Following rituximab therapy, longitudinal monitoring should be initiated from approximately month 4 onward, with monthly assessments to detect early plasmablast reconstitution that may precede clinical relapse [66,67,68]. Finally, plasmablast kinetics could be incorporated into retreatment decision-making, whereby exceeding an individualized plasmablast threshold, interpreted in conjunction with clinical and serological context, may help guide timely re-intervention and optimize disease control [69,70,102].
6.3. Digital Immune Phenotyping and Artificial Intelligence
The use of Artificial intelligence and machine learning approaches offer a powerful framework for integrating plasmablast kinetics with heterogeneous clinical and biological data streams to enhance flare prediction accuracy in autoimmune diseases [53,54]. By combining longitudinal plasmablast measurements with patient-reported outcomes and symptom quantification captured through mobile health applications, wearable-derived physiological and inflammatory signatures such as heart rate variability and activity patterns, and periodic laboratory data including conventional biomarkers and transcriptomic signatures, these models may detect subtle pre-flare states well before overt clinical or laboratory changes become apparent [42,53,54].
Such integrative, data-driven approaches hold the potential to shift autoimmune disease management toward truly preventive medicine, enabling anticipatory therapeutic adjustments days to weeks before clinical deterioration.
7. Future Directions in Plasmablast Research
The following directions reflect emerging research trajectories rather than established clinical practice.
7.1. Defining Plasmablast ‘Endotypes’
Just as asthma management has evolved from phenotype-based classification toward endotype-driven frameworks that enable targeted therapy selection, autoimmune diseases are increasingly moving toward molecular and immunological stratification [41,53,54]. In this context, plasmablast subsets defined through multi-omics profiling may delineate biologically distinct disease endotypes with direct therapeutic implications. Interferon-high endotypes, characterized by plasmablasts exhibiting strong type I interferon transcriptional signatures, are particularly prominent in systemic lupus erythematosus and Sjögren’s disease and may preferentially respond to interferon pathway blockade [42,43,44,45,46].
Oligoclonal IgG4 endotypes, defined by clonally restricted IgG4-producing plasmablast populations, underpin the pathogenesis of IgG4-related disease and highlight the antigen-driven nature of this condition [55,56,92]. Finally, ANCA-reactive extrafollicular endotypes, marked by robust plasmablast involvement and heightened extrafollicular activation, characterize subsets of ANCA-associated vasculitis and provide a mechanistic rationale for targeted B-cell and plasma cell-directed therapies in these patients [57,58,59] (Figure 7).
7.2. From Biomarker to Therapeutic Target
Plasmablast-depleting strategies are emerging as a pivotal therapeutic approach for patients with severe and refractory autoimmune diseases, reflecting a paradigm shift toward direct targeting of antibody-secreting cell compartments [26,27,71,72]. Ongoing and upcoming clinical trials evaluating BCMA-targeted antibody–drug conjugates, anti-CD38 monoclonal antibodies, and chimeric antigen receptor (CAR) T-cell therapies are expected to substantially reshape treatment landscapes for refractory autoimmunity. However, several critical questions remain to be addressed, including the optimal depth and duration of plasmablast and plasma cell depletion required to achieve durable remission, strategies to mitigate infectious complications associated with profound humoral immune suppression, and approaches to preserve protective immunity while selectively eliminating autoreactive antibody-producing cells [26,27,71,72,119,122,123,124]. Addressing these challenges will be essential to safely translate plasmablast-directed therapies into routine clinical practice and to fully realize their potential within precision immunology frameworks.
7.3. Spatial Immunology and Tissue-Based Assessment
Emerging spatially resolved technologies, including imaging mass cytometry, multiplexed ion beam imaging, and spatial transcriptomics, are providing unprecedented insight into the tissue distribution and functional context of plasmablasts within affected organs in autoimmune diseases [49,54,60]. In Sjögren’s disease, these approaches have revealed plasmablast clustering within periductal regions and ectopic lymphoid structures of salivary glands, supporting a role for local antibody production in glandular pathology [129,130]. In lupus nephritis, tubulointerstitial plasmablast infiltration has been linked to adverse renal outcomes, highlighting the pathogenic relevance of tissue-resident antibody-secreting cells beyond circulating biomarkers [49,60,63]. Similarly, in rheumatoid arthritis, plasmablasts localize within germinal center-like structures in inflamed synovium, reinforcing the concept that ectopic lymphoid microenvironments sustain localized humoral immune responses and contribute to chronic tissue inflammation [51,52,110].
7.4. Public Health and Health Economic Implications
Implementing plasmablast monitoring at scale has the potential to generate substantial public health benefits by enabling earlier and more precise intervention in autoimmune diseases. Anticipatory identification of impending flares could reduce cumulative glucocorticoid exposure through timely, targeted treatment adjustments, thereby limiting long-term toxicity. Preventing severe disease exacerbations before clinical deterioration may also decrease hospitalization rates and associated healthcare utilization. In parallel, biomarker-guided strategies could optimize medication use by supporting individualized dosing and retreatment schedules rather than fixed, time-based regimens, ultimately improving therapeutic efficiency while reducing unnecessary immunosuppression [102,103].
8. Conclusions
Plasmablasts represent a transformative biomarker at the interface of immunopathogenesis, precision immunology, and clinical decision-making [1,2,3,17,18]. Their rapid kinetics, mechanistic relevance, and predictive value position them as a cornerstone of next-generation autoimmune disease management. As short-lived progeny of activated B cells, plasmablasts provide real-time insight into ongoing immune dysregulation that cannot be obtained from traditional serological monitoring [8,9,10,11,12].
Across systemic lupus erythematosus, SjD, IgG4-related disease, ANCA-associated vasculitis, and rheumatoid arthritis, plasmablast dynamics correlate with disease activity, predict flares, and guide therapeutic decisions [19,55,57,73,104]. The kinetics of plasmablast reconstitution after B-cell–depleting therapy emerges consistently as one of the most powerful predictors of relapse, often surpassing traditional biomarkers in prognostic accuracy [66,67,68,116].
Advances in single-cell technologies have revealed remarkable heterogeneity within the plasmablast compartment, with distinct molecular subsets associated with specific diseases and pathogenic mechanisms [41,53,54]. These insights support endotype-based classification approaches that could guide precision therapy selection. The emergence of plasmablast- and plasma cell-directed therapies including anti-CD38 antibodies and BCMA-targeted agents provides new tools for patients with refractory disease [26,27,71,72].
Several key findings from this review have direct therapeutic implications. First, plasmablast kinetics following B-cell depletion can guide individualized retreatment timing, potentially reducing both under-treatment (premature relapse) and over-treatment (unnecessary immunosuppression). Second, the identification of interferon-high plasmablast subsets supports rational selection of interferon-blocking agents for appropriate patient populations. Third, the persistence of plasmablasts in SjD despite rituximab highlights the need for therapies with enhanced tissue penetration or alternative targets in this condition. Fourth, the remarkable responses to BCMA-targeted CAR-T cells in refractory SLE suggest that deep depletion of antibody-secreting cells may achieve effects beyond those attainable with conventional B-cell depletion. Finally, recognition that plasmablasts participate in cytokine networks—both responding to and producing inflammatory mediators—provides rationale for combination strategies targeting multiple nodes of immune dysregulation.
Implementation challenges remain, including standardization of measurement approaches, establishment of reference intervals, and integration into clinical workflows [31,32,33,119,121]. Nevertheless, the accumulating evidence supports plasmablasts as biomarkers ready for translation from research into clinical practice.
As multi-omic technologies mature and therapeutic targeting improves, plasmablast-guided medicine may evolve into a standard component of personalized care—moving from reactive treatment of established flares to proactive immune monitoring that enables preventive intervention. The shift from static serology to kinetic precision immunology represents a paradigm change with the potential to reduce organ damage, optimize therapy, and improve outcomes for patients with autoimmune diseases.
In summary, plasmablasts represent dynamic, context-dependent biomarkers that capture real-time immune activation across systemic autoimmune diseases. Their greatest clinical value lies not in isolated measurements but in longitudinal monitoring integrated with disease-specific and therapy-specific frameworks. While emerging plasmablast-targeted therapies hold promise, their translation into routine care will require careful patient selection, standardized monitoring, and long-term safety evaluation.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Nutt S.L. Hodgkin P.D. Tarlinton D.M. Corcoran L.M. The generation of antibody-secreting plasma cells Nat. Rev. Immunol.20151516017110.1038/nri 379525698678 · doi ↗ · pubmed ↗
- 2Shlomchik M.J. Weisel F. Germinal center selection and the development of memory B and plasma cells Immunol. Rev.2012247526310.1111/j.1600-065X.2012.01124.x 22500831 · doi ↗ · pubmed ↗
- 3Tellier J. Nutt S.L. Plasma cells: The programming of an antibody-secreting machine Eur. J. Immunol.201949303710.1002/eji.20184751730273443 · doi ↗ · pubmed ↗
- 4Shapiro-Shelef M. Calame K. Regulation of plasma-cell development Nat. Rev. Immunol.2005523024210.1038/nri 157215738953 · doi ↗ · pubmed ↗
- 5Victora G.D. Nussenzweig M.C. Germinal centers Annu. Rev. Immunol.20224041344210.1146/annurev-immunol-120419-02240835113731 · doi ↗ · pubmed ↗
- 6Cyster J.G. Allen C.D.C. B cell responses: Cell interaction dynamics and decisions Cell 201917752454010.1016/j.cell.2019.03.01631002794 PMC 6538279 · doi ↗ · pubmed ↗
- 7Mac Lennan I.C. Germinal centers Annu. Rev. Immunol.19941211713910.1146/annurev.iy.12.040194.0010018011279 · doi ↗ · pubmed ↗
- 8Ellebedy A.H. Jackson K.J.L. Kissick H.T. Nakaya H.I. Davis C.W. Roskin K.M. Mc Elroy A.K. Oshansky C.M. Elbein R. Thomas S. Defining antigen-specific plasmablast and memory B cell subsets in human blood after viral infection or vaccination Nat. Immunol.2016171226123410.1038/ni.353327525369 PMC 5054979 · doi ↗ · pubmed ↗