Genome-Wide Identification of RTE Gene Family Members in Sweet Potato and Their Expression Patterns Under Salt and Drought Stress
Xiaojie Jin, Heping Wan, Feng Yu, Xinsun Yang, Rongchang Yang

TL;DR
This study identifies and analyzes RTE genes in sweet potato, revealing their potential role in helping the plant adapt to salt and drought stress.
Contribution
The study reports the first genome-wide identification and expression analysis of RTE genes in sweet potato under abiotic stress.
Findings
Twenty-three RTE genes were identified in sweet potato, distributed across 21 chromosomes and one scaffold.
Most IbRTE genes showed tissue-specific and differential expression under drought and salt stress.
qRT–PCR confirmed that 14 representative IbRTE genes have distinct expression patterns under salt and drought treatments.
Abstract
Ethylene is a multifunctional phytohormone that regulates plant growth, development, and responses to abiotic/biotic stresses. RTE1 (Reversion-To-Ethylene Sensitivity1) acts as a negative regulator of the ethylene responses in Arabidopsis by positively regulating ethylene receptor ETR1. However, the role of RTE genes in sweet potato (Ipomoea batatas), an import food crop worldwide, remains largely unknown, particularly their involvement in abiotic stress adaptation. In this study, we identified 23 RTE genes in sweet potato, distributed across 21 chromosomes and one scaffold BrgTig00017944. The phylogenetic analysis divided them into two groups, the RTE1 group and RTH (RTE1-Homolog) group. Synteny analysis revealed that whole genome duplication (WGD) was the major force of expansion of the IbRTE gene family. Multiple cis-acting elements responsive to hormones and stress were found in the…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
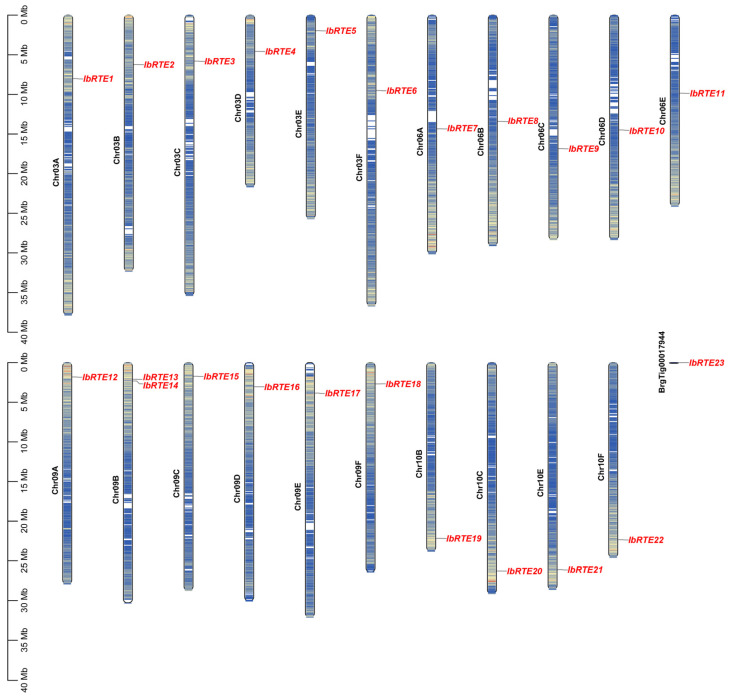 Figure 1
Figure 1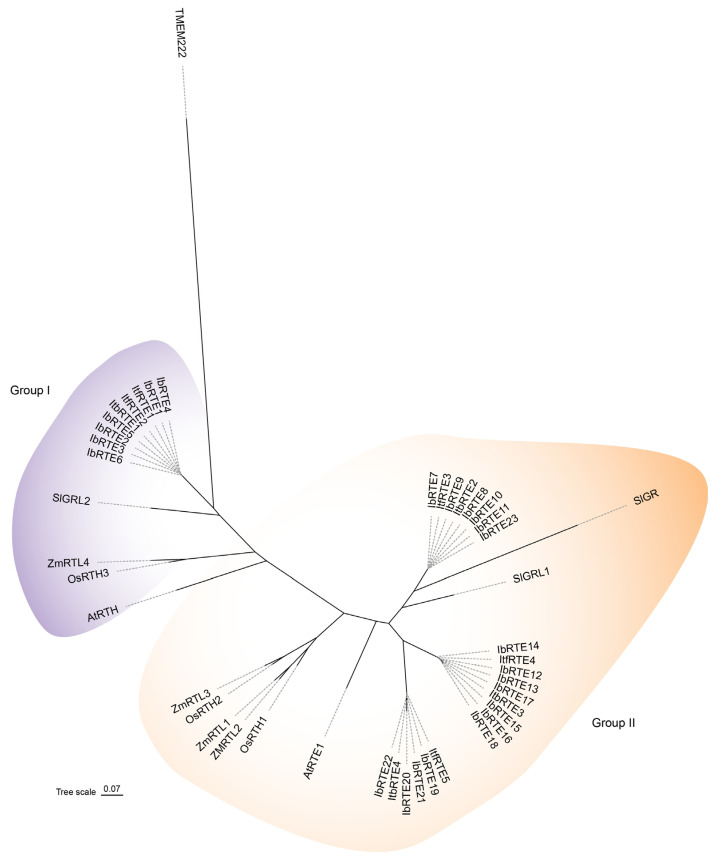 Figure 2
Figure 2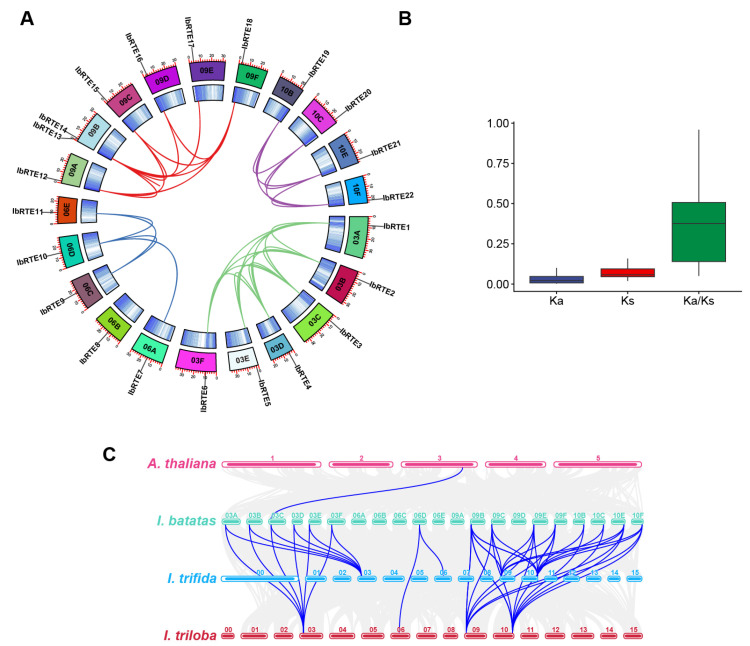 Figure 3
Figure 3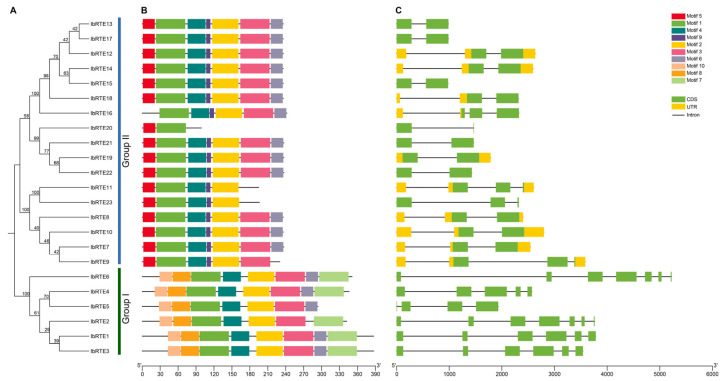 Figure 4
Figure 4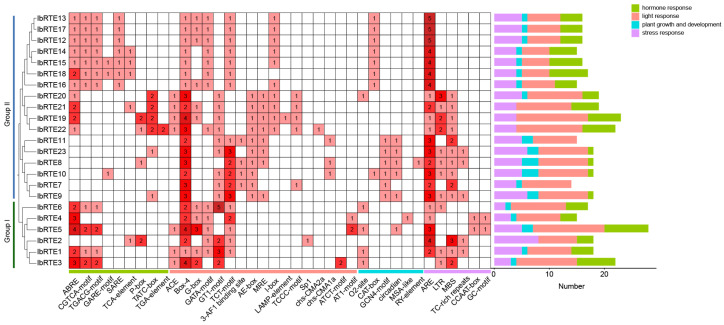 Figure 5
Figure 5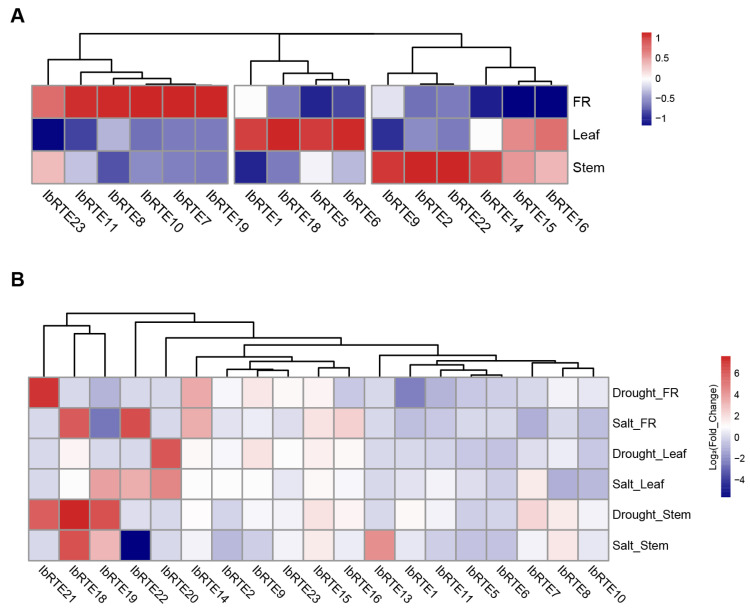 Figure 6
Figure 6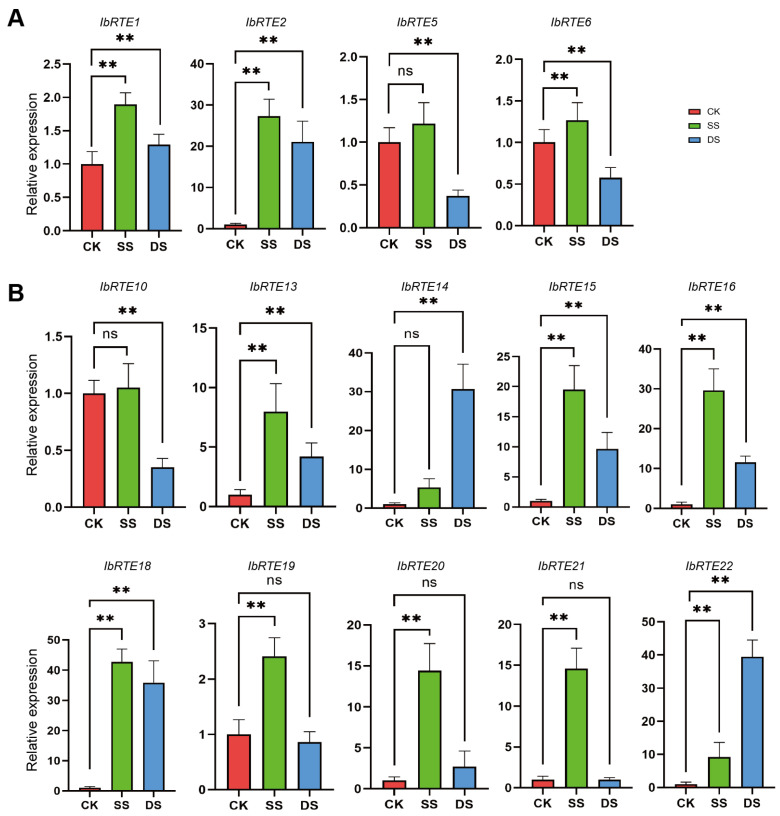 Figure 7
Figure 7- —Funding for seed industry high-quality development of Hubei Province
- —Technology Innovation Talents and Services Project of Hubei Province
- —CARS-10-Sweetpotato
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsPlant responses to water stress · Postharvest Quality and Shelf Life Management · Plant Stress Responses and Tolerance
1. Introduction
Sweet potato (Ipomoea batatas [L.] Lam) is a dicotyledonous plant belonging to the Convolvulaceae family and is recognized as one of the most important root crops globally, with annual world production approaching 100 million tons [1,2]. The storage roots, as the primary harvested organ, are rich in carbohydrates, dietary fiber, vitamins, and minerals and contain a variety of health-promoting bioactive compounds such as carotenoids, anthocyanins, flavonoids, and phenolic acids [3,4]. Due to its high nutritional value, usage versatility, and resilience, sweet potato contributes significantly to food security, animal feed, industrial applications, and bioenergy production [5,6]. It is widely cultivated in over 100 countries and is mainly grown on marginal land [7]. Therefore, it is necessary to improve its salt and drought tolerance to cope with the yield losses caused by the drought and salinity of marginal land [7].
Drought is one of the most frequent and devastating abiotic stresses, characterized by unpredictability, long duration, and wide-ranging impacts on plant physiology and yield [8]. It induces multiple physiological and biochemical responses, including stomatal closure, reduced photosynthesis, accumulation of reactive oxygen species (ROS), and altered hormone signaling [8,9]. Salt stress is another major abiotic constraint that leads to drought stress, ion toxicity (especially from Na^+^ and Cl^−^), and nutrient uptake imbalances, thereby impairing plant growth and productivity [10]. To ensure stable yields under adverse conditions, a better understanding of the genetic basis of stress tolerance in sweet potato is essential. Among the major hormonal pathways involved in stress response, the ethylene signaling pathway plays a central role [11].
Ethylene is a multifunctional phytohormone that regulates numerous developmental processes, including seed germination, hypocotyl elongation, root hair formation, leaf senescence, abscission, fruit ripening, and responses to both abiotic and biotic stressors [12,13]. Ethylene signaling operates through a well-coordinated pathway in which ethylene receptor-interacting RTE proteins can modulate ethylene signaling by interacting with ethylene receptors and influencing downstream components, thereby fine-tuning plant physiological responses and environmental adaptability [13,14,15]. RTE1 of Arabidopsis was isolated from a suppressor screen of the dominant ethylene-insensitive mutant etr1-2 [16]. The AtRTE1 protein co-localizes and interacts directly with AtETR1, a key ethylene receptor, in the Golgi and endoplasmic reticulum, modulating a negative feedback loop on ethylene response adaptation [13,16,17]. The RTE1 gene family is highly conserved in plants, animals, and protists. In Anadiplosis, RTH (RTE1-Homolog) is the only homolog of the RTE1 gene family with 61.5% similarity, and acts via RTE1 in regulating ethylene response and signaling [16,18,19]. In rice, three RTE1 homologous genes have been identified. Among them, OsRTH1 and OsRTH2 are clustered in the same clade as AtRTE1, while OsRTH3 is grouped with AtRTH. However, only OsRTH1 is involved in regulating ethylene responses in rice. Overexpression of OsRTH1 significantly suppresses ethylene-induced developmental changes, including leaf senescence, seedling leaf elongation, coleoptile elongation and curvature, and adventitious root development [18]. In maize, six ZmRTE genes have been identified, and most exhibit higher expression in leaves under salt stress, suggesting a role in shoot-based stress adaptation [20]. Additionally, ZmRTL2 and ZmRTL4 interact with members of the ARGOS protein family in maize to modulate ethylene signaling, thereby reducing ethylene sensitivity and enhancing drought tolerance and grain yield under stress conditions [21,22]. Similarly, in cotton, GhRTE6 has been shown to be involved in both developmental processes and salt stress responses, underscoring the diverse functionality of RTE genes [23].
Despite the extensive studies in other species, no comprehensive genome-wide investigation of the RTE gene family has been reported in sweet potato. The species is an autohexaploid (2n = B_1_B_1_B_2_B_2_B_2_B_2_ = 6X = 90) with a large and highly heterozygous genome [24]. This genomic complexity results in intricate genetic traits, which have hindered gene discovery through forward genetics approaches [24,25]. Nevertheless, it also presents unique opportunities for investigating novel gene functions and evolution processes. Therefore, a systematic analysis of RTE genes in sweet potato is needed to elucidate their structure, evolutionary relationships, and potential functional roles under stress conditions.
In this study, we identified 23 IbRTE genes in the sweet potato genome and conducted comprehensive analyses of their gene structures, conserved motifs, promoter cis-acting elements, and phylogenetic relationships. We also assessed their expression profiles under drought and salt stress using transcriptome and qRT–PCR. Our findings contribute to a better understanding of the roles of RTE genes in ethylene-mediated stress responses and provide a theoretical basis for breeding in stress-tolerant sweet potato.
2. Materials and Methods
2.1. Plant Materials and Stress Treatments
Sweet potato cultivar Eshu 11 was used as the experimental material for this study. Stem cuttings (~25 cm in length) with apical tips were inserted into the planting medium at a depth of ~10 cm. The cuttings were placed in Hoagland nutrient solution supplemented with either 20% PEG-6000 (w/v) or 200 mM NaCl to simulate drought and salt stress, respectively, with five plants per treatment. An additional five cuttings were placed in Hoagland solution without PEG or NaCl and used as the control group. The third unfolded leaves were collected at 7 days after planting from three random plants per treatment, representing three biological replicates. The samples were immediately flash-frozen in liquid nitrogen and stored at −80 °C until RNA extraction. All experiments were conducted in a greenhouse at the School of Life Sciences, Jianghan University, Wuhan, Hubei, China (30°30′ N, 114°9′ E). All treatment and control plants were arranged on the same shelf level, following a completely randomized block design.
2.2. Identification of IbRTE Genes and Analysis of Physicochemical Properties
The genome sequences of both autohexaploid sweet potato (Ipomoea batatas [L.] Lam.) and two diploid sweet potatoes (Ipomoea triloba and Ipomoea trifida) were downloaded from their genome database (http://sweetpotato.uga.edu/, accessed on 18 December 2025). The RTE1 and RTH (RTE1-Homolog) proteins of Arabidopsis thaliana were retrieved from TAIR (https://www.arabidopsis.org/, accessed on 18 December 2025) and used as queries to identify RTE genes in sweet potato by local BLASTP(version 2.17.0) searches. In parallel, the hidden Markov model (HMM) profile of the RTE domain (PF05608) was downloaded from the Pfam database (http://pfam.xfam.org/, accessed on 18 December 2025), and HMMER (http://www.hmmer.org/, accessed on 18 December 2025) was used to search for RTE-containing sequences in the sweet potato genome (E < 1 × 10^−10^) [26]. The results from both methods were combined, redundant sequences were removed, and candidate genes were further confirmed using the NCBI-CDD (https://www.ncbi.nlm.nih.gov/cdd/, accessed on 14 September 2025) and Pfam databases [26,27]. The physicochemical properties of the IbRTE proteins, including amino acid length, molecular weight (MW), isoelectric point (pI), instability index, aliphatic index, and grand average of hydropathicity (GRAVY), were analyzed using the ProtParam tool on ExPASy (https://web.expasy.org/protparam/, accessed on 14 September 2025) [28]. Then, subcellular localization of the IbRTE proteins were predicted using WoLF PSORT (https://wolfpsort.hgc.jp/, accessed on 18 December 2025) [29].
2.3. Sequence Alignment and Phylogenetic Analyses
RTE/RTH protein sequences from Arabidopsis thaliana, Oryza sativa, Zea mays, Ipomoea triloba, Ipomoea trifida, and Ipomoea batatas were aligned using Muscle, and a phylogenetic tree was constructed using the Maximum Likelihood method in IQ-TREE [30], with 1000 bootstrap replicates. The resulting phylogenetic tree was visualized using the web application ChiPlot (https://www.chiplot.online/, accessed on 18 December 2025) [31].
2.4. Gene Structure, Conserved Domain, and Protein Motif Analysis
The exon/intron structure information of IbRTEs was extracted from the genome annotation file. Conserved domains were confirmed using the NCBI-CDD and Pfam databases [26,27], and conserved motifs were identified using MEME (https://meme-suite.org/, accessed on 18 December 2025) with the motif number set to 10 [32]. Results were visualized using TBtools [33].
2.5. Chromosomal Localization and Synteny Analysis
The chromosomal positions of the IbRTE genes were retrieved from the genome annotation file, and their distribution was mapped using TBtools. The collinearity relationships within the Ipomoea batatas genome, as well as among the genomes of Ipomoea batatas, Arabidopsis thaliana, Ipomoea triloba, and Ipomoea trifida, were identified using MCScanX v1.0.0, and visualized by TBtools v2.311 [33,34]. The Ka (non-synonymous substitutions), Ks (synonymous substitutions), and Ka/Ks values of the IbRTE gene pairs were calculated using TBtools.
2.6. Cis-Acting Element Analysis
The 2000 bp genomic sequence upstream of the translation initiation site of each IbRTE was extracted from the genome data and analyzed for cis-acting elements using PlantCARE (https://bioinformatics.psb.ugent.be/webtools/plantcare/html/, accessed on 14 September 2025) [35]. Based on their functions, the cis-acting elements associated with hormone response, light response, plant growth and development, and stress response were analyzed.
2.7. Expression Profile Analysis
The RNA-seq data of Xushu 18 under drought, salt and normal conditions were downloaded from NCBI (accession PRJNA511028). After quality control, the clean reads were aligned to the sweet potato genome by HISAT2 [36]. Gene expression levels were calculated in FPKM using StringTie [37]. The expression profiles of IbRTEs in the fibrous root, stem, and leaves under the three treatments were analyzed and visualized by R package pheatmap v1.0.13.
2.8. qRT–PCR Validation of IbRTE Expression
Total RNA was extracted from sweet potato leaves under drought and salt stress using the FastPure Universal Plant Total RNA Isolation Kit (Vazyme, Nanjing, China). First-strand cDNA was synthesized with the HiScript^®^ II Q Select RT SuperMix with a gDNA wiper (Vazyme, Nanjing, China). Fourteen representative IbRTEs were selected based on their cis-acting element types and transcriptome data, and their relative expressions in leaves were validated by qRT-PCR. Specific primers for the qRT–PCR were designed using Primer Premier 5 and the β-Actin gene was used as the internal reference (Table S1). qRT–PCR was performed on an AriaMx Real-Time PCR System (Agilent Technologies Inc., Santa Clara, CA, USA) using the following conditions: 95 °C for 30 s, followed by 40 cycles of 95 °C for 5 s, and 55 °C for 1 min. The relative expression levels were calculated using the 2^–ΔΔCT^ method [38], with three biological and three technical replicates per sample.
3. Results
3.1. Identification and Characterization of the IbRTE Genes in Sweet Potato
By means of combinate BLASTP(version 2.17.0) research and HMMER research to the sweet potato genome, a total of 23 IbRTEs were identified in the sweet potato genome, and all these IbRTEs contain the conserved domains of DUF778 or the DUF778 superfamily. The genes were named IbRTE1–IbRTE23 and anchored to their respective chromosomes according to their chromosomal location (Figure 1). IbRTE23 was located on the scaffold BrgTig00017944, and its chromosomal position could not be precisely determined. The remaining genes were unevenly distributed across chromosomes 3, 6, 9, and 10. Chromosome 9 contained the largest number of genes (seven genes), followed by chromosomes 3 (six genes) and 6 (five genes), while chromosome 10 contained the fewest (four genes).
The physicochemical properties analysis revealed that the IbRTE genes encode proteins ranging from 98 to 386 amino acids in length, with molecular weights (MWs) between 11.08 and 44.35 kDa and theoretical isoelectric points (pI) ranging from 5.22 to 7.08. The aliphatic index varied from 82.05 to 102.32. Notably, IbRTE1–IbRTE6 had an instability index > 40, indicating that these proteins may be unstable, whereas the rest had an instability index < 40, suggesting relative stability. The average hydropathicity (GRAVY) values indicated that five IbRTE proteins were hydrophilic, owing to their negative scores, whereas the remaining proteins were predicted to be hydrophobic. These results demonstrate substantial variation in the physicochemical properties of the 23 IbRTE proteins (Table 1). In addition, subcellular localization prediction results showed that most RTE-encoded proteins in sweet potato are localized to either the plasma membrane (16) or the cytoplasm (6). Notably, IbRTE20 was localized to the nucleus.
3.2. Phylogenetic Analysis of IbRTEs
To explore the evolutionary relationship of IbRTEs, the homologous protein sequences from Ipomoea batatas, Arabidopsis thaliana, Oryza sativa, Zea mays, Ipomoea trifida, and Ipomoea triloba were aligned (Figure S1, Table S2), and a phylogenetic tree was constructed using the Maximum Likelihood (ML) method. The results revealed that IbRTE1-IbRTE6, OsRTH3, ZmRTL4, and AtRTH (AT3G51040) were clustered in group I, suggesting that they are RTE1-homologous genes (Figure 2). Meanwhile, IbRTE7-IbRTE23, OsRTH1, OsRTH2, ZmRTL1-ZmRTL3, three ItfRTEs, and three ItbRTEs were in group II with AtRTE1 (AT2G26070), suggesting that their roles are similar to AtRTE1. The sequence identity and similarity results showed that IbRTE1-IbRTE6 had higher identity with and similarity to AtRTH, while other IbRTEs had higher identity with and similarity to AtRTE1 (Table S3). In addition, the sequence alignments show that IbRTE proteins in the same chromosome group have the most similarity, and IbRTE1-IbRTE6 in the third chromosome group have the lowest similarity to the other IbRTE proteins (Figure S1). IbRTE23, which was located on a scaffold, has 98.97% similarity to IbRTE 11, which was located on chromosome 6E.
3.3. Synteny Analysis of IbRTEs
Intra-genomic collinearity analysis was conducted to explore the gene duplications among the 23 IbRTE genes. A total of 35 collinear gene pairs within the IbRTE family were found. Among them, only one gene pair on Chr09B, IbRTE13-IbRTE14, was identified as a tandemly duplicated gene pair. Meanwhile, 34 gene duplication events involving 21 IbRTEs were identified. No gene duplication in IbRTE8 was found (Figure 3A). Interestingly, gene duplication events only occurred between IbRTEs in the same chromosome group, including 14 pairs in chromosome 3, 4 pairs in chromosome 6, 11 pairs in chromosome 9, and 6 pairs in chromosome 10. Some IbRTEs have collinearity with 2-5 other IbRTE genes, such as IbRTE1 (5), IbRTE2 (4), and IbRTE14 (4), etc., suggesting shared structural and functional characteristics. In addition, the Ka/Ks values of the collinear gene pairs were all less than one, indicating that the IbRTEs were subject to purifying selection pressure (Figure 3B, Table S4).
To further explore the phylogeny and evolutionary history of IbRTE genes, synteny analysis between sweet potato and three other species, including A. thaliana, I. trifida, and I. triloba, was conducted (Figure 3C). There are 1, 16, and 16 IbRTEs (IbRTE1-IbRTE6, IbRTE10, IbRTE13-IbRTE15, and IbRTE17-IbRTE22) that show synteny with A. thaliana, I. trifida, and I. triloba, respectively (Figure 3C). Among them, only IbRTE3 showed a syntenic relationship with AtRTH of Arabidopsis. A total of 23 syntenic gene pairs were found between 16 IbRTEs of I. batatas and 4 ItfRTEs of I. trifida (ItfRTE2, ItfRTE3, ItfRTE4, and ItfRTE5). ItfRTE2 had a collinear relationship with IbRTE1-IbRTE6, while both ItfRTE4 and ItfRTE5 showed collinearity with seven IbRTEs (IbRTE13-IbRTE15, IbRTE17, IbRTE18, IbRTE21, and -IbRTE22) on chromosomes 9 and 10. Similarly, 23 syntenic gene pairs were found between 16 IbRTEs of I. batatas and 4 ItbRTEs of I. triloba (ItbRTE1, ItbRTE2, ItbRTE3, and ItbRTE4). ItbRTE1, ItbRTE3, and ItbRTE4 displayed a collinear relationship with multiple IbRTE genes. These results reflect closer genome conservation and a closer evolutionary relationship among I. batatas, I. trifida, and I. triloba.
3.4. Gene Structure, Conserved Motifs, and Cis-Acting Element Analysis
The conserved motif analysis revealed that Motif 1 was present in all IbRTE proteins, and Motif 4, Motif 2, and Motif 3 were present in the majority (Figure 4A,B). In group I, all IbRTE proteins possessed Motif 10 and Motif 8, and most of them contained Motif 7, suggesting that these motifs may be associated with specific functions. Conversely, genes in group II lacked Motif 10, Motif 8, and Motif 7 but possessed Motif 5 and Motif 9. The exon–intron structure analysis showed that the number of exons ranges from two to seven. Genes in group II generally contained fewer exons than those in group I, including two exons (seven), three exons (seven), and four exons (three). Each gene in group I had more than four exons, and IbRTE2 and IbRTE6 had the largest number of exons (seven) (Figure 4A,C).
To explore the potential functions and regulatory mechanisms of the IbRTEs, the cis-acting elements in the 2000 bp upstream promoter regions were analyzed. The prediction results revealed a rich abundance of elements related to light, stress, and hormonal responses, highlighting the potential role of the IbRTE genes in responding to environmental and hormonal cues (Figure 5). The stress response elements of IbRTEs are mainly associated with anaerobism (ARE), low-temperature (LTR), and drought (MBS), indicating that IbRTEs may be involved in various stress-response processes. Notably, IbRTE12-IbRTE18 on chromosome 9 only contain the ARE element, suggesting their potential role in anaerobic induction responses. In addition, various types of hormone-responsive elements were predicted, including an abscisic acid (ABA)-responsive element (ABRE), jasmonic acid (JA)-responsive elements (CGTCA-motif and TGACG-motif), gibberellin (GA)-responsive elements (GARE-motif, P-box, and TATC-box), salicylic acid (SA)-responsive elements (SARE and TCA-element), and an auxin-responsive element (TGA-element). These hormones were closely associated with plant stress resistance. Therefore, the IbRTE genes may be widely involved in hormone-regulated stress-response pathways.
3.5. Expression Patterns of IbRTE Genes Under Salt and Drought Stress
To investigate the functional roles of these IbRTE genes in response to abiotic stress, the transcriptome data of fibrous roots, leaves, and stems under normal, drought, and salt conditions were downloaded from NCBI with the accession number PRJNA511028, and analyzed. As shown in Figure 6, 16 IbRTEs were detected in three tissues under all three conditions. IbRTE20 was expressed under both drought and salt stresses, while IbRTE21 and IbRTE13 were expressed under drought and salt stress, respectively. Under normal conditions, the expression of IbRTEs can be roughly classified into three patterns, including highly expressed in the fibrous root (six), leaf (four), and stem (six) (Figure 6A). Compared with normal treatment, most IbRTEs were up-regulated under drought and salt treatment, especially IbRTE14, IbRTE15, IbRTE16, and IbRTE18 (Figure 6B). Therefore, these four IbRTEs were speculated to play roles in sweet potato’s responsiveness to drought and salt stresses.
To validate the IbRTE expressions, qRT-PCR was conducted to evaluate the expression profiles of 14 representative IbRTE genes under salt (NaCl, 200 mM) and drought (PEG, 20% W/V) treatments (Figure 7). All four IbRTEs (IbRTE1, IbRTE2, IbRTE5, and IbRTE6) in group I were up-regulated under salt stress, especially IbRTE1, IbRTE2, and IbRTE6, with significant differences. They were also significantly changed under drought stress, with two up-regulated (IbRTE1 and IbRTE2) and two down-regulated (IbRTE5 and IbRTE6). Among the ten IbRTEs in group II, the vast majority of them were significantly up-regulated under both salt and drought stresses. Notably, five IbRTEs in the 9th chromosome, particularly IbRTE13, IbRTE15, IbRTE16, and IbRTE18, were highly up-regulated under both abiotic stresses, suggesting their potential roles in the response to both salt and drought stresses.
4. Discussion
As ethylene receptor-interacting proteins, RTE genes negatively regulate ethylene responses and signaling in plants [13,14,15]. While the regulatory role of individual RTE genes has been well-documented in Arabidopsis, rice, maize, tomato, and other species [16,17,18,19,20,21,22,23,39,40,41], genome-wide identification and expression profiling across the RTE gene family remain limited in sweet potato (Ipomoea batatas). In this study, we identified 23 IbRTE genes of sweet potato (Figure 1), a number substantially higher than that reported in other species, including Arabidopsis (two), maize (six) and upland cotton (eight) [16,20,23]. Gene duplication represents a major force driving the expansion and evolution of gene families [42]. Sweet potato has a complex autohexaploid genome, and a whole genome duplication is considered to have occurred after crosses between progenitors [24]. The intra-genomic synteny analysis revealed that 34 paralogous pairs were found (Figure 3A, Table S4), suggesting that whole genome duplication might be the major force driving the expansion of the IbRTE gene family. In addition, only one tandem duplication was found, suggesting a low frequency of tandem duplication events in the expansion of the IbRTE gene family. Moreover, the Ka/Ks value of all IbRTE gene pairs were less than one (Figure 3B), indicating that they underwent purifying selection during the evolution and domestication of sweet potato. This selective pressure likely contributes to the functional conservation of the IbRTE gene family.
Consistent with previous phylogenetic analysis, phylogenetic analysis across Ipomoea batatas, Arabidopsis thaliana, Oryza sativa, Zea mays, Ipomoea trifida, and Ipomoea triloba grouped the IbRTEs into two major groups, the RTE1 group and RTH group [18,39,41]. Among them, IbRTE1-IbRTE6, OsRTH3, SlGRL2, and AtRTH were clustered together, whereas IbRTE7-IbRTE23, OsRTH1, OsRTH2, SlGR, and SlGRL1 were in the same group (Figure 2). Genes within the same group generally exhibited similar exon–intron structures and conserved motifs (Figure 4), indicating functional conservation. Previous studies reveal that AtRTH and SlGRL2 play roles distinct from RTE1 in ethylene signaling [19,41]. Therefore, it is speculated that IbRTE1-IbRTE6 may have similar functions to AtRTH, while IbRTE7-IbRTE23 may have similar functions to AtRTE1.
Previous studies in Arabidopsis and tomato have revealed that RTE1 and its homologs, GR and GRL1, regulate ethylene receptor activity, and thereby modulate ethylene sensitivity [40,41,43]. The overexpression of AtRTE1/SlGR/SlGRL1 genes in these systems can enhance ethylene receptor activity, reducing sensitivity to ethylene and influencing processes such as fruit ripening and senescence [16,19,40,41]. Furthermore, the interaction of RTE1 with ARGOS (auxin-regulated gene involved in organ size) proteins has been shown to modulate plant organ size and contribute to drought tolerance, possibly through crosstalk between the ethylene and auxin signaling pathways [21,22]. In sweet potato, promoter analysis of IbRTE genes revealed the presence of multiple cis-regulatory elements responsive to ABA, JA, GA, SA, and auxin, among which the most abundant elements are those related to ABA and JA (Figure 5). Both ABA and JA are crucial hormones in mediating various plant stress defense responses [44,45]. Furthermore, IbRTEs are enriched in stress-responsive elements, including ARE, LTR, and MBS. This suggests that IbRTEs may integrate into complex hormonal and environmental signaling networks and potentially participate in stress-inducible transcriptional programs. Interestingly, a disproportionately large number of light-responsive elements were distributed among the promoters. Among them, Box 4 is present in all IbRTEs promoters, while TCT-motif, GT1-motif, and G-box are also widely present (Figure 5). Ethylene is one of the main inducers for the breakdown of chlorophyll, which causes leaf senescence [46]. This emphasizes the possible involvement of IbRTEs in light-regulated developmental processes, such as photomorphogenesis or chlorophyll metabolism, which remain largely unexplored for this gene family.
To better understand the expression pattern of IbRTEs, their expression profiling via RNA-seq in fibrous roots, leaves, and stems under three treatments (normal, drought, and salt) were analyzed. As shown in Figure 6, 16 IbRTEs were tissue-specially expressed under normal conditions, while 18 and 18 IbRTEs were expressed under drought and salt stress, respectively. Compared to normal conditions, IbRTE20 was expressed under both stresses, while IbRTE21 and IbRTE13 were expressed under drought and salt stress, respectively. Combining the results of cis-acting element analysis and transcriptome analysis, the expression profiling via qRT-PCR of 14 representative IbRTEs under salt and drought stresses supports the functional diversification of IbRTE genes in abiotic stress responses (Figure 7). The majority of them were up-regulated in response to abiotic stresses, particularly under salt stress (11 genes), mirroring trends observed in other species like maize and cotton [20,23]. Notably, IbRTE19, IbRTE20, and IbRTE21 were significantly up-regulated under salt stress but exhibited stably expression under drought stress. Similarly, IbRTE10 and IbRTE14 were stable expressed under salt stress, but significantly down- and up-regulated under drought stress, respectively. This differential regulation pattern suggests that different IbRTE members may play distinct roles, either as primary responders or as modulators of stress response pathways. However, further functional studies are required to confirm their precise roles in response to abiotic stresses.
Taken together, these results highlight the significant role of the RTE gene family in sweet potato’s stress tolerance and adaptive growth. The combined evidence from genome-wide characterization, phylogenetics, promoter analyses, and expression studies provides a solid foundation for future functional studies, shedding light on the regulatory mechanisms by which RTE genes help plants cope with salt and drought stress.
5. Conclusions
In this study, a genome-wide investigation of the RTE family in sweet potato identified 23 IbRTE genes successfully mapped to 21 sweet potato chromosomes, with the exception of IbRTE23 being unanchored. Phylogenetic and structural analyses classified these genes into two distinct groups, with highly conserved motifs and structural elements suggesting potential functional similarities within each group. Synteny analysis indicated that the vast majority of IbRTEs arose from whole genome duplication events. Numerous cis-regulatory elements’ responses to stress and hormones were identified in the putative promoter regions of IbRTEs. The expression profiling revealed that IbRTE genes have tissue-specific and independent expression patterns under drought and salt stresses. These findings advance our understanding of the structure, evolution, and expression patterns of the RTE family in sweet potato and provide valuable theoretical and genetic resources for future studies.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Behera S. Chauhan V.B.S. Pati K. Bansode V. Nedunchezhiyan M. Verma A.K. Monalisa K. Naik P.K. Naik S.K. Biology and biotechnological aspect of sweet potato (Ipomoea batatas L.): A commercially important tuber crop Planta 20222564010.1007/s 00425-022-03938-835834064 · doi ↗ · pubmed ↗
- 2Food and Agriculture Organization of the United Nations (FAO)Available online: https://www.fao.org/faostat/en/(accessed on 7 May 2025)
- 3Wang A. Li R. Ren L. Gao X. Zhang Y. Ma Z. Ma D. Luo Y. A comparative metabolomics study of flavonoids in sweet potato with different flesh colors (Ipomoea batatas (L.) Lam)Food Chem.201826012413410.1016/j.foodchem.2018.03.12529699652 · doi ↗ · pubmed ↗
- 4Tanaka M. Ishiguro K. Oki T. Okuno S. Functional components in sweet potato and their genetic improvement Breed. Sci.201767526110.1270/jsbbs.1612528465668 PMC 5407917 · doi ↗ · pubmed ↗
- 5Sapakhova Z. Raissova N. Daurov D. Zhapar K. Daurova A. Zhigailov A. Zhambakin K. Shamekova M. Sweet potato as a key crop for food security under the conditions of global climate change: A review Plants 202312251610.3390/plants 1213251637447081 PMC 10346279 · doi ↗ · pubmed ↗
- 6Tang C. Li M. Jiang B. Ejaz I. Ameen A. Mo X. Zhi M. Wang Z. High-throughput profiling of sweet potato vine biomass for cellulosic ethanol production using near-infrared spectroscopy and chemometrics Microchem J.202521311367910.1016/j.microc.2025.113679 · doi ↗
- 7Zhang H. Gao X. Zhi Y. Li X. Zhang Q. Niu J. Wang J. Zhai H. Zhao N. Li J. A non-tandem CCCH-type zinc-finger protein, Ib C 3H 18, functions as a nuclear transcriptional activator and enhances abiotic stress tolerance in sweet potato New Phytol.20192231918193610.1111/nph.1592531091337 · doi ↗ · pubmed ↗
- 8Razi K. Muneer S. Drought stress-induced physiological mechanisms, signaling pathways and molecular response of chloroplasts in common vegetable crops Crit. Res. Biotechnol.20214166969110.1080/07388551.2021.187428033525946 · doi ↗ · pubmed ↗