The Perspective of Using Ischemic Tolerance in Clinical Practice
Rastislav Burda, Marián Sedlák, Jozef Burda

TL;DR
This paper explores using ischemic tolerance, a natural protective mechanism, to treat ischemia-reperfusion injuries in clinical settings.
Contribution
It proposes transferring ischemic tolerance via activated blood plasma from healthy donors to patients unable to build their own tolerance.
Findings
Ischemic tolerance effectors can be transferred through blood plasma.
Activated plasma may treat ischemia-reperfusion injuries in patients unable to self-generate tolerance.
Experimental evidence suggests this method could be effective in clinical practice.
Abstract
Ischemic–reperfusion injury represents an extremely serious problem in the human population. It mainly affects the elderly population and currently used treatments have poor results. However, in nature there is a much more effective and relatively well-studied mechanism known as the ischemic tolerance phenomenon. If an organism is exposed to adverse conditions that do not destroy it, it responds by producing substances capable of protecting it from severe damage or death in the event of a repeated encounter with the same or a different dangerous environment. The problem with its use in the clinic is that its effectiveness decreases in the elderly and is practically lost with associated diseases and their concurrent treatment. Based on experimental animal studies and findings, it can be assumed that the activation of full tolerance—through successive exposure to two stressors in young,…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
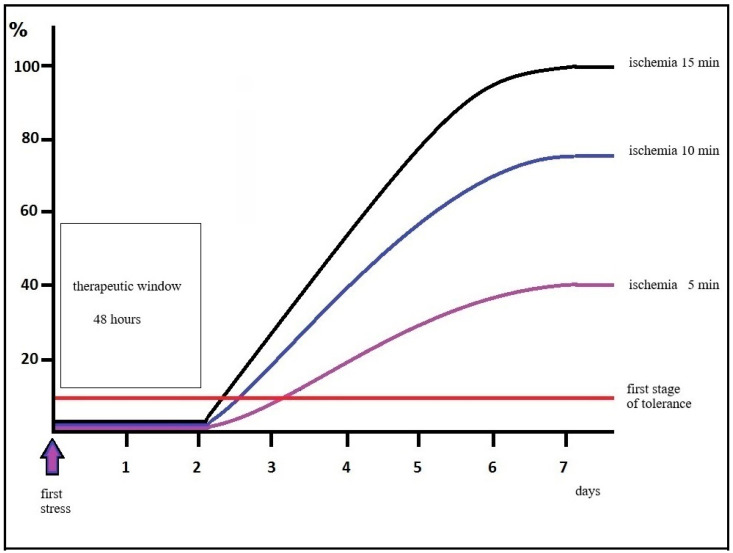 Figure 1
Figure 1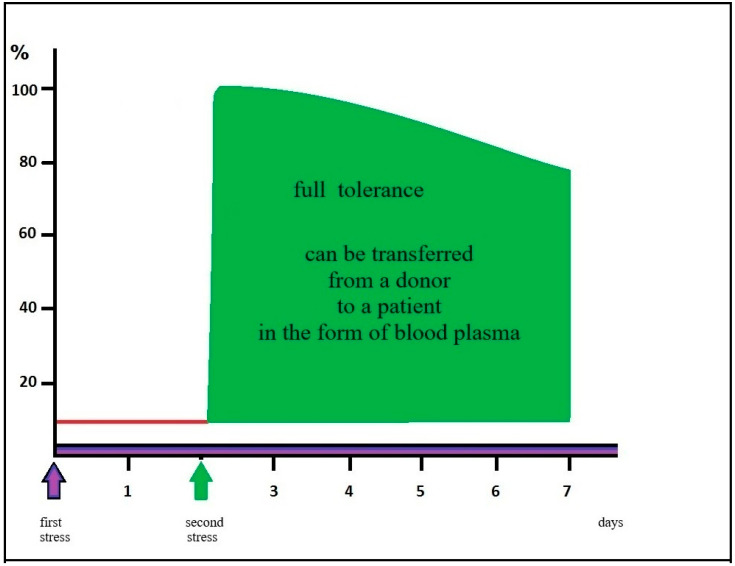 Figure 2
Figure 2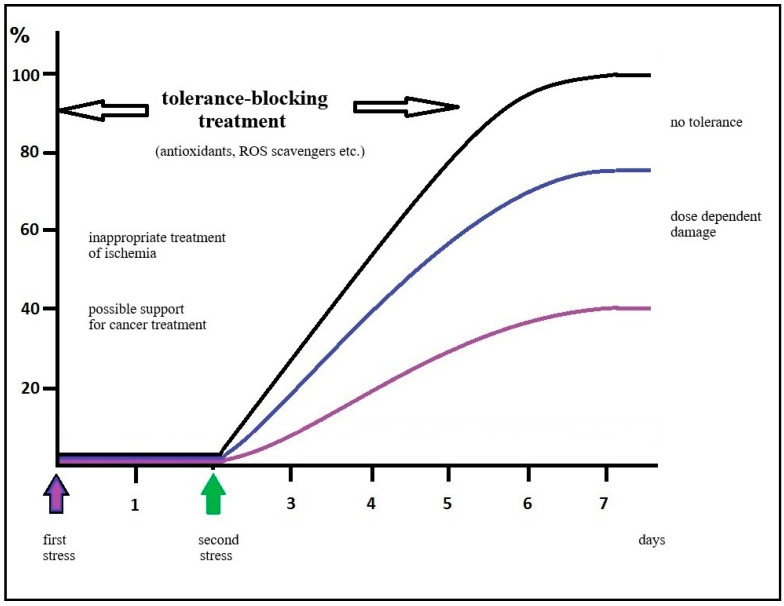 Figure 3
Figure 3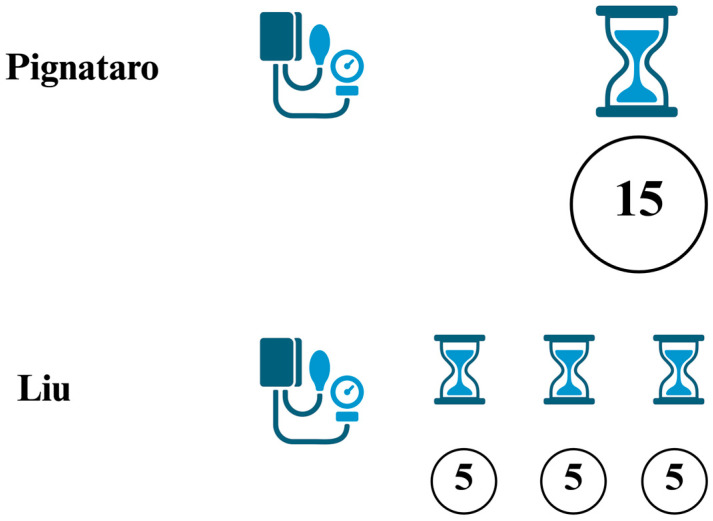 Figure 4
Figure 4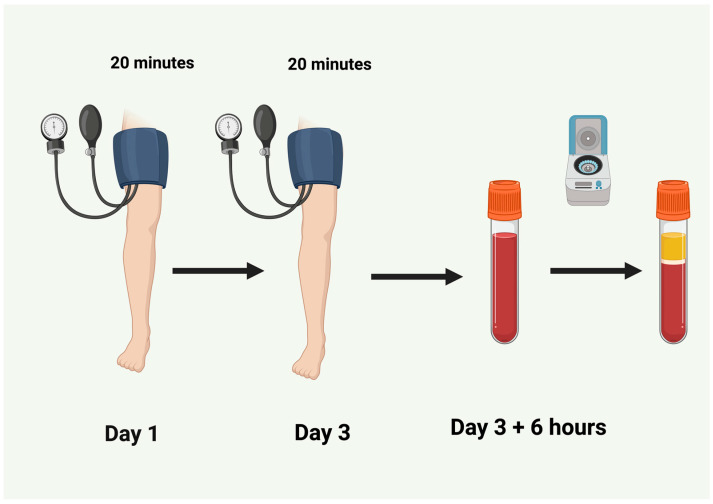 Figure 5
Figure 5Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsCardiac Ischemia and Reperfusion · Organ Transplantation Techniques and Outcomes · Trauma, Hemostasis, Coagulopathy, Resuscitation
1. Introduction
Living organisms in nature possess a rare and ultimately essential attribute: a relentless drive for survival. Even in the most sensitive population of cells or organisms, a certain percentage of resistant cells are usually found in the face of the same insult that proves fatal for most. This “miracle of nature” is known as ischemic tolerance. The first scientific evidence was presented by Murry and colleagues [1].
Establishing this protective phenotype in response to stress depends on a coordinated response at the genomic, molecular, cellular, and tissue levels [2].
If an organism is exposed to adverse conditions that do not destroy it, it reacts by producing substances capable of protecting it from severe damage or death in the event of repeated contact with a dangerous environment. The classic saying “what doesn’t kill you makes you stronger” is, in fact, true and holds under certain conditions. This ability is referred to as ischemic tolerance. Fascinatingly, this also works in reverse: if an organism or part of it finds itself in a potentially devastating situation, repeated similar stress may help it survive. Hence, the saying “a wedge drives out a wedge” is also applicable [3].
In addition, this mechanism is remarkable in that one type of stimulus can induce resistance to other types of stimuli—this is known as “cross-tolerance”. It may explain why individuals subjected to appropriate hypothermia (such as ice swimmers) exhibit resistance to various other forms of damage. Another valuable aspect of this mechanism is the so-called “remote tolerance”—the ability to induce protection across the entire organism, even when the initial stimulus affects only a part [4]. This is enabled by the protective substances of tolerance that circulate in the bloodstream [5].
In this review, we summarize the available experimental and clinical data of effectiveness of ischemic tolerance. It can be expected that ischemic tolerance can be used in the treatment of ischemic–reperfusion injury. The current partial knowledge of the mechanisms of ischemic–reperfusion injury does not provide the possibility of a comprehensive treatment of this multifactorial injury, while based on the available experimental works it should be expected that effectors of ischemic tolerance from “active” plasma could be used in the treatment of ischemic injury until the molecular identification of the effectors occurs.
2. Pathophysiology of Ischemia–Reperfusion Injury
Ischemia induces multifactorial changes in the organism, which are usually aggravated by changes caused by subsequent reperfusion. The cessation or strong reduction of oxygen supply, essential for aerobic oxidative phosphorylation in mitochondria, leads to a decrease in ATP. This causes a malfunction of Na^+^/K^+^-ATPase, and due to an increased influx of Na^+^ and water, the cell swells. The cell switches to anaerobic glycolysis, resulting in the accumulation of lactic acid and a decrease in pH [6].
Failure of both Na^+^/K^+^-ATPase and Ca^2+^-ATPase is manifested by potassium leakage from the cell and calcium accumulation in the cytoplasm, which leads to the activation of proteases and phospholipases. Mitochondrial damage and opening of the mitochondrial pore (MPTP) indicate cytochrome c leakage and the initiation of apoptosis [7]. If ischemia lasts too long, cell death occurs, mostly in the form of uncontrolled necrosis. Mild damage may lead to programmed cell death—apoptosis. In addition to necrosis as the resulting state of cell death, various states and processes leading to different forms and modes of cell death can be observed, and necrosis itself does not occur uniformly but in many ways (apoptosis, mitopsosis, necrosis, necroptosis, apoptosis, and delayed neuronal death) [8,9,10].
Ischemia leads initially to necrosis, but following reperfusion accelerates apoptosis, which occurs in almost 86% of cell death after prolonged ischemia [11,12,13,14].
Postischemic reperfusion causes complex cellular changes that may result in further damage (reperfusion lesion) or protective adaptations (in the case of conditioning). In the first minutes of reperfusion, so-called reactive hyperemia usually occurs. Tissue weakened by ischemia is unable to process the sudden oxygen influx, creating conditions for the formation of free radicals [15]. Explosive production of reactive oxygen species (ROS) is driven by the activity of NADPH oxidase, xanthine oxidase, and the mitochondrial electron transport chain [16,17]. The result is damage to lipids, proteins, and DNA [18].
Excessive accumulation of Ca^2+^ and ROS due to the opening of the mitochondrial permeability transition pore (mPTP) leads to decreased ATP synthesis and, in more severe cases, can result in apoptosis or necrosis [19,20]. During ischemia and reperfusion, protein synthesis is also significantly affected, although postconditioning can modulate this process in favor of cell survival. The cessation of protein synthesis during ischemia is a consequence of energy collapse. In the first minutes of reperfusion, ribosomes disintegrate from mRNA, stopping protein synthesis [21]. Disaggregation of polyribosomes is caused by phosphorylation of eIF2α, which blocks the formation of the translation initiation complex [22].
Reperfusion also allows protein synthesis to resume, but often with pathological consequences, such as the synthesis of pro-apoptotic proteins (e.g., Bax) [23]. At the same time, the accumulation of misfolded proteins in the endoplasmic reticulum activates the UPR (unfolded protein response) via the IRE1α, PERK, and ATF6 pathways [24,25].
In summary, it can be stated that during IR damage following pathological reaction occur: intracellular Ca and Na accumulation with rapid pH change, loss of mitochondrial membrane potential, occurrence of oxidative stress, massive ROS formation and uric acid generation, ROS-induced ROS generation, changes in NO metabolism, endothelial dysfunction, cytokines and chemokine signaling changes, expression of cell adhesion molecules, neutrophil tissues infiltration, platelet aggregation, and microembolization leading to autophagy and apoptosis [26].
3. Treatment Strategy of IR Injury
Many ways to reduce the extent of IR damage have been described so far. In recent years, attention has been paid to NO protective strategy (adenosine), [27] inhibition of apoptosis [28] and Ca^2+^ excess in the cell [29], antioxidants (SOD, catalase, N-ACC, vitamin E [30],) Na^+^H^+^ channel inhibitors [31], glutamate depletion [32,33], controlled reperfusion/reoxygenation [34], intermittent ischemia, neutrophil cell depletion [35], Aprotinin [36] poly (ADP-ribose) polymerase (PARP inhibitors) [37], blockade of the complement system, anesthetics, hypothermia, and hyperbaric oxygen therapy.
Currently, most attention is paid to the following strategies:
3.1. Pharmacological Agents
Glycogen synthase kinase-3β (GSK-3β) inhibitors is a multifunctional serine/threonine kinase which is involved in a variety of biological processes, including cell proliferation, apoptosis, and immune response. Inhibition or inactivation of GSK-3β provides protection against IR injury, making it a viable target for drug development. Though numerous GSK-3β inhibitors have been identified to date, the development of therapeutic treatments remains a considerable distance away. It is a potential target for future IRI therapy [38].
c-Jun N-terminal kinase (JNK) inhibitors has redox-centric mechanism, and its activation is involved in cell proliferation, migration, and invasion while inducing apoptosis and G1-phase cell cycle arrest. Its activation also has a future potential for treatment of brain tumor metastasis [39].
3.1.1. Xanthine Oxidase Inhibitors
Allopurinol reduced oxidative stress which was the result of hypoxia/hyperoxia, as shown by decreased 8-isoprostane plasma concentration. However, during hypoxia, as well as hyperoxia, allopurinol administration resulted in a significant increase in autonomic control upon the heart as shown by increased standard deviation of all normal NN intervals with an increased vagal contribution [40]. Xanthine Oxidase Inhibitors have a promising effect in reducing oxidative stress in cardiac, lung, and brain cells [41], but their therapeutic potential in IRI remains under future investigation.
3.1.2. Trimetazidine
Trimetazidine inhibits long-chain 3-ketoacyl-CoA thiolase; it changes energy reliance from fatty acid oxidation to glucose metabolism, and it reduces inflammation and the extent of IR damage [42].
3.1.3. GSK-3β Inhibitors
GSK-3β inhibitors represent an adaptive response that might limit the extent of adverse remodeling in the aftermath of acute myocardial infarction, promote angiogenesis and reduce myocardial remodeling, especially after acute myocardial infarction, decrease apoptosis and fibrosis, and are very effective in tissue repair [43].
3.1.4. JNK Inhibitors
JNK pathways are involved in diverse cellular processes, including growth regulation, transformation, and programmed cell death, and they are closely linked with cerebral IR injury [44], so inhibitors like SP600125 is promising for future therapy [45].
3.2. Anti-Inflammatory Therapies
3.2.1. NF-κB Inhibitors
NF-κB is a transcription factor, which plays a role in inflammatory response during IR injury, and is involved in the release of pro-inflammatory factors and apoptosis of cardiomyocytes [46].
3.2.2. RAGE (Receptor for Advanced Glycation End Products) Inhibitors
RAGE inhibition leads to lower expression of cytokines, which is responsible for endothelial dysfunction [47].
3.3. Regenerative Medicine
3.3.1. Exosome-Based Therapy
Exosomes are membrane-bound extracellular vesicles (EVs) that are produced in the endosomal compartment of most eukaryotic cells. Exosomes derived from adipose-derived MSCs protect ischemic myocardium from I/R injury through the activation of Wnt/β-catenin signaling pathway [48]. Compared to MSCs, MSC-derived exosomes reveal many advantages such as non-immunogenicity, easy access, easy preservation, and extreme stability under various conditions [49]. Its usage is promising for future application [50].
3.3.2. Micro RNA
MicroRNA, or rather some clusters, such as miR-17~92, can be highly expressed in endothelial cells during ischemia. Using targeted mRNA antagonists, better angiogenesis and functional repair of damaged tissue can be achieved in experiments [51].
None of the above-mentioned therapies have yet found significant use in practice.
4. Discussion—Ischemic Tolerance as a Therapeutic Strategy
Ischemic tolerance is a phenomenon where a sublethal ischemic insult protects against a subsequent, more severe ischemic event. This protective effect is an intrinsic mechanism where cells “learn” to tolerate stressful conditions.
Conditioning is a strategy whose goal is activation of ischemic tolerance. It should be applied as a brief interruption of blood flow, applied before (preconditioning), or during the early phase of reperfusion following an ischemic event (postconditioning). The onset of ischemia is unexpected; therefore, the use of preconditioning is very limited in clinical settings.
If ischemia–reperfusion changes are multifactorial, it logically follows that the treatment must also offer multifactorial protection. This is where the potential use of postconditioning arises. Compared to unprotected reperfusion, which itself causes further damage, postconditioning induces significant beneficial changes at the cellular and molecular levels.
Postconditioning significantly attenuates ischemia-induced neuronal death, suppresses the release of MnSOD and cytochrome c, and prevents caspase-3 activation [7,52]. It stabilizes the mitochondrial membrane through Akt/ERK activation and GSK-3β inhibition, preventing the opening of the mPTP [53]. It simultaneously increases the expression of Bcl-2 and HSP70, thereby blocking apoptotic pathways [54]. Postconditioning also enhances NO production via eNOS and improves vasodilation [55]. Experimental studies show that it also reduces the extent of damage in cerebral ischemia [56,57,58]. Moreover, a 5 min interruption of blood flow to the brain in a laboratory rat causes the death of up to 40% of the most sensitive CA1 hippocampal neurons, and 10 min without oxygen leads to the destruction of up to 70% of these cells. However, if a 10 min ischemia is applied within the right time window after a 5 min ischemia, there is no accumulation of damage; on the contrary, fewer than 10% of neurons die. This means that the combination of two otherwise lethal stimuli is required to save the cells [56,59].
Several important points must be noted here: after 5 or even 10 min of ischemia, not all brain cells die—and certainly not immediately. Even the most sensitive nerve cells in the brain, namely the neurons of the first layer of Ammon’s horn in the hippocampus, begin to die only after approximately 48 h, through a process known as delayed neuronal death [8,9]. This suggests that there is a substantial therapeutic window during which these cells can still be rescued. If each of these stresses can independently destroy 40% and 70% of neurons, respectively, their combination—when timed optimally—activates tolerance mechanisms that save the cells. Moreover, it does not matter whether the stronger or weaker stimulus is applied first.
Preconditioning, but especially postconditioning, can prevent the death of the most sensitive neurons in the brain (CA1 hippocampus), even at durations of oxygen deprivation exceeding the 5 min threshold, which is otherwise lethal without conditioning. Until recently, the 5 min limit was considered the point at which irreversible changes begin to develop. Our results show that CA1 hippocampal neurons can be rescued even after delayed postconditioning applied 48 h after a lethal 10 min brain ischemia [56,60,61]. This represents a therapeutic window of several hours to days in cases of reversible ischemia or hypoxia of the brain or the whole body.
The delayed postconditioning effect described in the CA1 hippocampus is graphically illustrated in the following Figure 1 and Figure 2.
The effect of “remote conditioning” has come to the fore, where the stressor needs to be applied to only part of the body—most often a limb—to induce the effect. Stopping blood flow using an inflatable cuff for a single period of several minutes or through several repeated short (30 s to 5 min) periods of ischemia interrupted by equally long reperfusion times induces sufficient tolerance. Brief episodes of limb ischemia are safe and applicable to a wide range of potential organ damage, including both ischemic and toxic injury [62,63,64].
During the last three decades, a wide variety of ischemic conditioning strategies and pharmacological treatments have been tested in the clinic. However, their translation from experimental to clinical studies for improving patient outcomes has been both challenging and disappointing [65]. Moreover, preclinical trials often involve young, healthy animals, which do not account for factors like aging or other chronic conditions that significantly impact the outcomes in human patients [26,66].
The results of human studies have been mixed, with some showing no effect (beta-blockers, remote ischemic conditioning [67], Cangrelor [68]), and some showing promising results (colchicine [69], interleukin 1 blockade [70], Canakinumab [71]). In most cases, it was only a partial influence on one of the complex mechanisms of IR damage, not a comprehensive treatment of IR damage.
Sex hormones and sex-specific signaling pathways also play an important role in modulating their efficacy in the presence of comorbidities on animal models, though some findings indicate that comorbidities can differentially affect IRI between sexes; this field remains till now underexplored. Most preclinical studies focus on healthy animals, often neglecting sex and comorbidity influences and very often neglecting the combination of sex and comorbidities.
Incorporating both sexes and relevant comorbidities in experimental models is essential for a comprehensive understanding of biological responses to IR injury [72].
Several problems can arise when applying conditioning. Activation of tolerance can be prevented using antioxidants [73], proteasome inhibitors [74], or other substances such as naloxone [75], and high glucose concentrations also prevent the development of tolerance [76]. On the other hand, the use of antioxidants—by preventing the development of tolerance—could significantly interfere with the treatment of diseases where we aim to eliminate unwanted cells from the body.
Metabolic syndrome (MetS), a cluster of conditions including abdominal obesity, dyslipidemia, hypertension, metainflammation, and glucose intolerance, represents a multi-pronged assault on cardioprotective mechanisms. The effect of conditioning decreases with age and with the presence of diseases, including diabetes [77,78], high cholesterol [79], and obesity [80].
Based on the mentioned results, use of antioxidants, protein synthesis inhibitors, or inhibitors of specific enzyme activity can prevent the development of tolerance; on the other hand, preventing (blocking) the activation of tolerance, which inevitably occurs during cancer treatment, could significantly enhance the outcomes of this treatment (Figure 3).
Remote conditioning shows that there is no need to develop tissue-specific conditioning methods (e.g., neuro-conditioning or cardio-conditioning). A comparison of conditioning methods seems more favorable for remote conditioning, practiced by applying an inflatable cuff to the limb. Here, the question arises whether to use one longer interval of ischemia, as described by Pignataro [81], or to apply more (most often three) cycles of shorter intervals interspersed with roughly equal reperfusion intervals, as described by Liu [82] (Figure 4).
The essential condition for the use of postconditioning is that it must be applied in a way that is both safe and acceptable for the patient (i.e., the patient can tolerate the tourniquet application time). Postconditioning can be used at any time after lethal stress; it can also be applied repeatedly over several days and, of course, can be combined with other treatment procedures. A method that reliably induces tolerance in elderly, ill, and medicated patients would be invaluable.
Another important problem is the reproducibility of the results of individual studies, which is why the IMPACT studies represent a significant challenge. Numerous cardioprotective interventions have been reported to reduce myocardial infarct size (IS) in pre-clinical studies. However, their translation for the benefit of patients with acute myocardial infarction (AMI) has been largely disappointing. One reason for the lack of translation is the lack of rigor and reproducibility in pre-clinical studies. Pig AMI multicenter European network with centralized randomization and core blinded IS analysis was established and validated with the aim to improve the reproducibility of cardioprotective interventions in pre-clinical studies and the translation of cardioprotection for patient benefit. It was performed in a similar way in another study on small animals (rodents and rabbits) [83,84].
5. Future Perspectives
Logically, further research continues to identify the key mechanisms and substances responsible for ensuring tolerance. On the other hand, it has been proposed to begin applying the knowledge gained so far, particularly regarding the spread of tolerance effectors via the humoral route (proofed in animal model) [5] and their transfer in the form of blood derivatives from young, healthy, unmedicated donors to generate neuroprotective plasma.
It can be expected that the triggers of ischemic tolerance are contained in plasma; they are probably low-molecular hydrophobic substances and are independent of local neurogenic activity and require activation of local opioid receptors [5].
Since the effectiveness of remote ischemic postconditioning has also been proven in the case of delayed neuronal death induced by temporary global brain ischemia or kainate intoxication (animal model), it can be assumed that the conditioning products are able to overcome the blood–brain barrier [62].
The effect of preconditioned plasma (activated plasma) was already demonstrated by Zhao [85]. Preconditioned plasma (collected from donor animals 48 h after limb ischemia) was able to reduce the extent of IR myocardial damage after transfer to recipients, which was clinically manifested by a reduction in the incidence and duration of ventricular tachycardia. A very similar result was also achieved by Weber [86], who by applying ischemic preconditioned plasma to volunteers achieved protection of human umbilical endothelial cells from hypoxia-induced damage. In the mentioned case, the plasma was taken away directly after finishing preconditioning. Weber and Zhao in all the above cases made only a transfer of “partially” activated plasma. The plasma was activated only by one stress, meaning that only ischemic tolerance triggers were present in it. On the other hand, in our experiments, double-activated plasma (exposure of animals to 2 stresses within a time of 48 h, plasma was collected 6 h after the second stress) was used. By collecting double-activated plasma, effectors, not ischemic tolerance triggers, should be included in plasma. The advantage of double-activated (“fully activated”) plasma containing complete activated ischemic tolerance also lies in the fact that comedication and comorbidity in the recipient will probably have no effect on its immediate effectiveness.
Unless there is a clear chemical identification of the effectors of ischemic tolerance, the simplest and easiest way to activate ischemic tolerance will be to prepare activated plasma from young healthy donors with the possibility of its immediate use in recipients during initial treatment. This does not exclude repeated administration and simultaneous use of other treatment methods.
Based on our experimental work on animal models [87,88,89], we recommend the following applicable timeline for the preparation of activated plasma, based on published work:
Day 1: Initial conditioning (e.g., a single 10–20 min limb ischemia session or three cycles of 5–8 min of cuff inflation/deflation). A subsequent 48 h period of “reperfusion” allows activation of the first stage of tolerance and accumulation of signaling molecules ready for a rapid response to the second stimulus [90].
Day 3: Second conditioning (using the same protocol as on Day 1). Blood collection follows 6 h, allowing time for synthesis or activation (via posttranslational modifications) of tolerance effectors (Figure 5).
The above-mentioned usage of activated plasma has not been performed in the clinical trials yet and is just a hypothetical scheme suggested by the authors.
Based on our experimental work on animal models, it is expected that blood plasma should be effective immediately after administration into the patient’s bloodstream, but it should also retain its activity after 30 days of storage at −80 degrees Celsius, as well as after lyophilization. It has been shown on animal models to be effective not only in cerebral ischemia but also in 3MT (3-methyltin) intoxication [87]. Due to its conditioning effect, it has also shown potential in protecting against other toxic substances such as kainate in the brain [60,62,64], and doxorubicin [91] and isoprenaline [92] in the heart and muscle.
6. Conclusions
Anoxic, ischemic, traumatic, and toxic brain damage represent a huge individual and family tragedy, as well as a big economic burden. The use of current therapy means long-term mild improvement with lifelong consumption of an expensive medication.
The difference between the treatments used in clinics and the potential use of ischemic tolerance is incomparable. Tolerance allows surviving multiple lethal doses, and it is a rapid and inexpensive technique.
While most of the methods used so far aim to mitigate the damage that has already occurred, ischemic tolerance prevents this damage by utilizing the phenomenon of delayed neuronal death.
The concept of “active plasma” holds enormous potential for emergency medicine and the prevention of neurodegeneration, and it may lead to the first universal neuroprotective therapy based on humoral factors.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Murry C.E. Jennings R.B. Reimer K.A. Preconditioning with ischemia: A delay of lethal cell injury in ischemic myocardium Circulation 1986741124113610.1161/01.CIR.74.5.11243769170 · doi ↗ · pubmed ↗
- 2Gidday J.M. Cerebral preconditioning and ischaemic tolerance Nat. Rev. Neurosci.2006743744810.1038/nrn 192716715053 · doi ↗ · pubmed ↗
- 3Zemke D. Smith J.L. Reeves M.J. Majid A. Ischemia and ischemic tolerance in the brain: An overview Neurotoxicology 20042589590410.1016/j.neuro.2004.03.00915474608 · doi ↗ · pubmed ↗
- 4Przyklenk K. Bauer B. Ovize M. Kloner R.A. Whittaker P. Regional ischemic ‘preconditioning’ protects remote virgin myocardium from subsequent sustained coronary occlusion Circulation 19938789389910.1161/01.CIR.87.3.8937680290 · doi ↗ · pubmed ↗
- 5Shimizu M. Tropak M. Diaz R.J. Suto F. Surendra H. Kuzmin E. Li J. Gross G. Wilson G.J. Callahan J. Transient limb ischaemia remotely preconditions through a humoral mechanism acting directly on the myocardium: Evidence suggesting cross-species protection Clin. Sci.200911719120010.1042/CS 2008052319175358 · doi ↗ · pubmed ↗
- 6Siesjo B.K. Katsura K.I. Kristian T. Li P.A. Siesjo P. Molecular mechanisms of acidosis-mediated damage Acta Neurochir. Suppl.19966681410.1007/978-3-7091-9465-2_28780790 · doi ↗ · pubmed ↗
- 7Danielisova V. Gottlieb M. Nemethova M. Kravcukova P. Domorakova I. Mechirova E. Burda J. Bradykinin postconditioning protects pyramidal CA 1 neurons against delayed neuronal death in rat hippocampus Cell. Mol. Neurobiol.20092987187810.1007/s 10571-009-9369-319259804 PMC 11505757 · doi ↗ · pubmed ↗
- 8Kirino T. Delayed neuronal death in the gerbil hippocampus following ischemia Brain Res.1982239576910.1016/0006-8993(82)90833-27093691 · doi ↗ · pubmed ↗