Multi-Omics Evidence Linking Depression to MASLD Risk via Inflammatory Immune Signaling
Keye Lin, Yiwei Liu, Xitong Liang, Yiming Zhang, Zijie Luo, Fei Chen, Runhua Zhang, Peiyu Ma, Xiang Chen

TL;DR
This study finds that depression increases the risk of liver disease through inflammation and identifies a potential immune target for treatment.
Contribution
The study provides multi-omics evidence of a causal link between depression and MASLD via inflammatory immune signaling, identifying the CD40LG-CD40 axis as a novel mechanism.
Findings
Depression significantly increases MASLD risk, particularly in women.
Inflammatory markers like hs-CRP, GGT, and ALP mediate the depression-MASLD relationship.
CD40LG is identified as a key molecular bridge between depression and MASLD via immune signaling.
Abstract
Background: Depression and Metabolic Dysfunction-Associated Steatotic Fatty Liver Disease (MASLD) are common chronic diseases, respectively. However, the causal and molecular links between them remain unclear. In order to explore whether depression contributes to an increased risk of MASLD and whether inflammation mediates this effect, we integrated multi-level evidence from the epidemiology of the National Health and Nutrition Examination Survey (NHANES), the genetics of GWAS, the transcriptomes of GEO, and single-cell RNA sequencing datasets. Methods: A multi-level integrative analysis strategy was used to validate this pathway. First, a cross-sectional epidemiological analysis based on NHANES data was used to reveal the association between depression and MASLD, and to explore the mediating role of inflammation and liver injury markers. Secondly, a two-sample Mendelian randomization…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
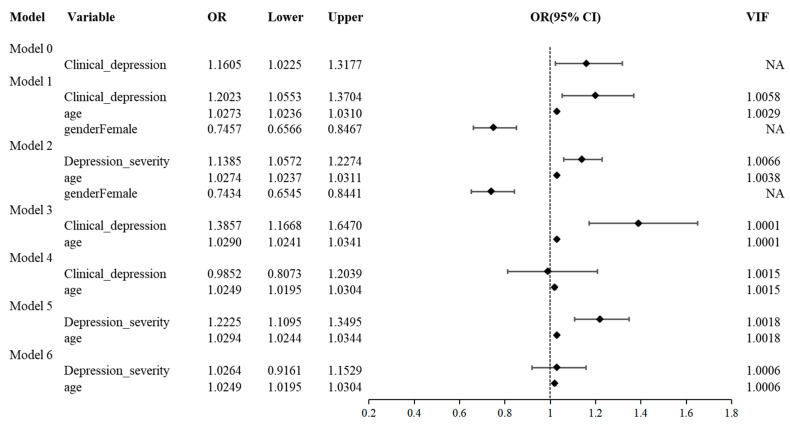 Figure 1
Figure 1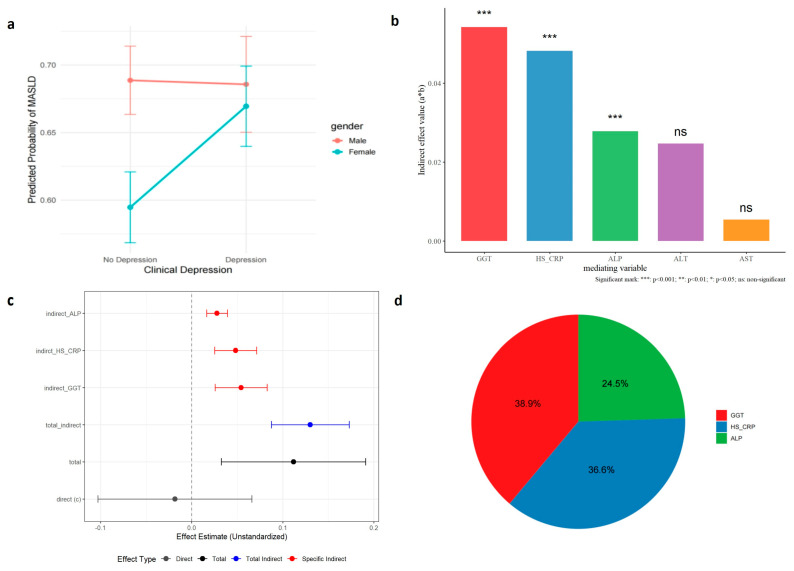 Figure 2
Figure 2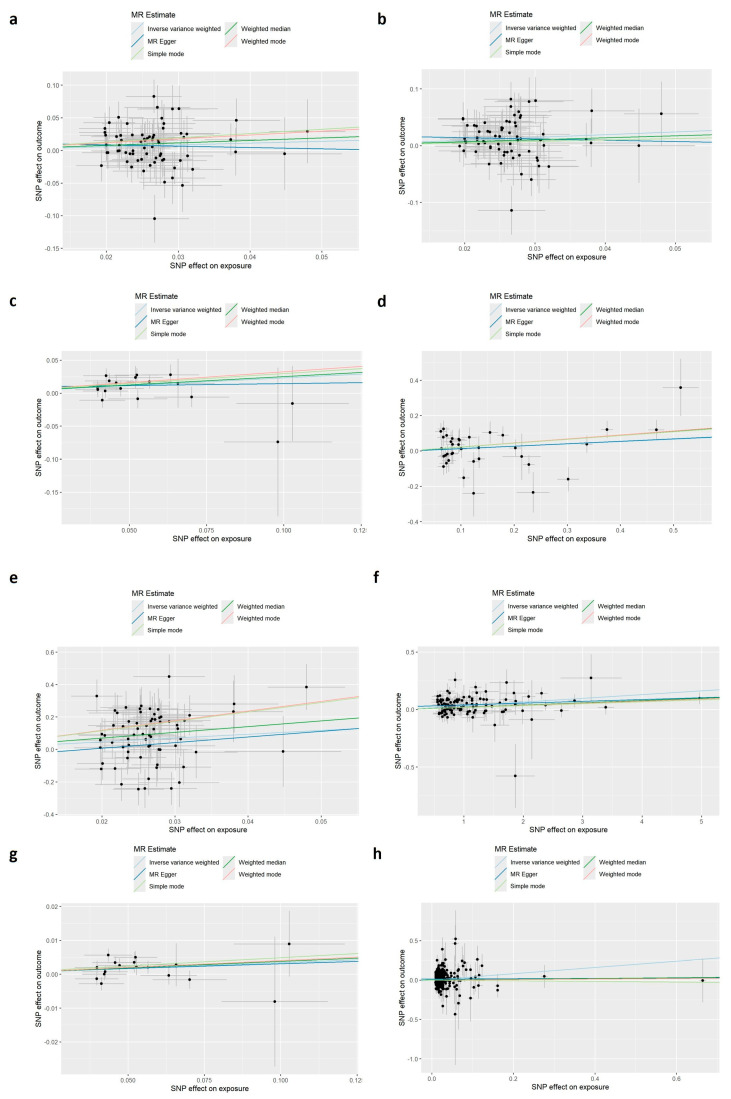 Figure 3
Figure 3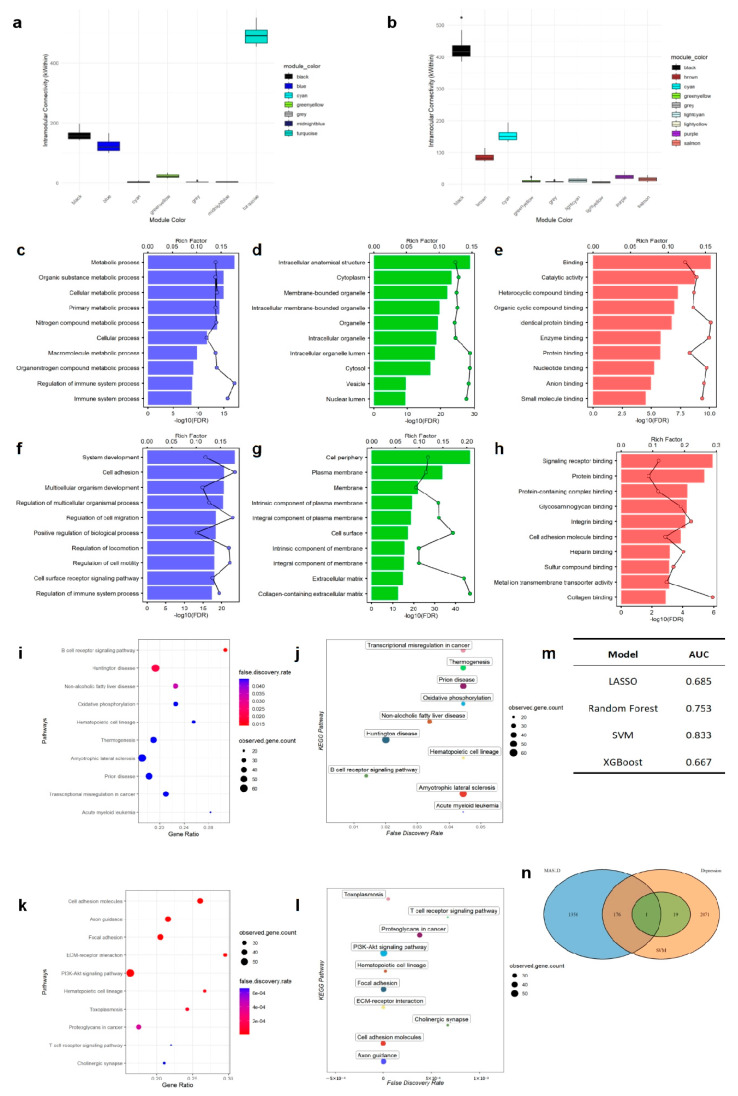 Figure 4
Figure 4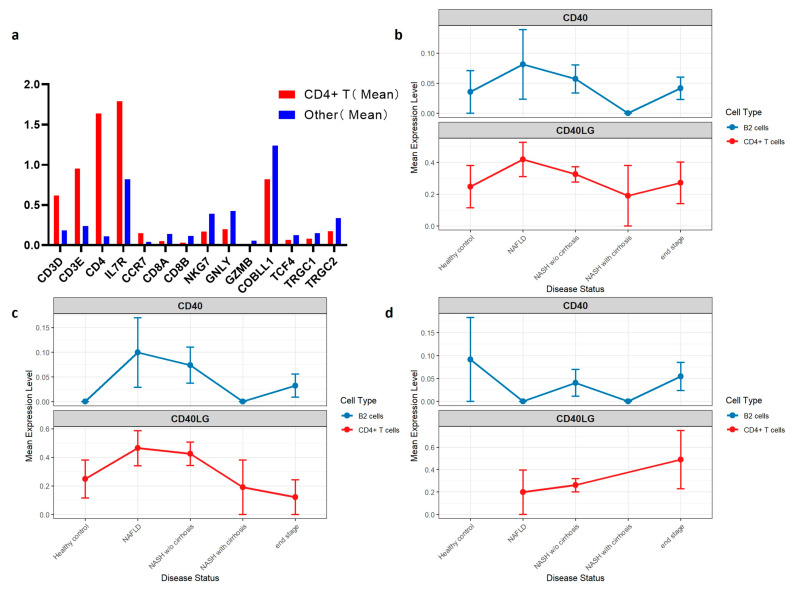 Figure 5
Figure 5- —Guangdong Basic and Applied Basic Research Foundation
- —Basic Research Program of Guangzhou Science and Technology Bureau Jointly funded Dengfeng Hospital Project
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsGenetic Associations and Epidemiology · Liver Disease Diagnosis and Treatment · Ferroptosis and cancer prognosis
1. Introduction
Depression is one of the most prevalent mental health disorders worldwide. According to the 2021 Global Burden of Disease (GBD) study, depression affects over 330 million people and accounts for more than 56 million disability-adjusted life years (DALYs), with consistently higher prevalence in females than in males [1]. Metabolic dysfunction–associated steatotic liver disease (MASLD), formerly known as nonalcoholic fatty liver disease (NAFLD), is the most common chronic liver disease globally. It affects over one-third of the adult population and contributes to more than 4 million DALYs [2,3]. Like depression, MASLD is a chronic, progressive condition whose prevalence has steadily risen over the past three decades. Notably, a recent study found that 26.3% of MASLD patients also have depression, indicating a substantial overlap between these conditions [4].
Accumulating evidence suggests that chronic, low-grade inflammation serves as a common pathophysiological platform linking depression and MASLD. Recent reviews emphasize that systemic inflammation is a key modifiable contributor to depression, with elevated cytokines and acute-phase proteins being hallmarks of this state [5]. On the other hand, MASLD is intrinsically linked to hepatic and systemic inflammation, and research by A. Teixeira et al. identified the Systemic Immune-Inflammation Index (SII) as a reliable and independent risk marker for the disease [6]. This forms a cross-organ inflammatory network that links central neuroinflammation to hepatic metabolic dysfunction. The research by E. F. Osimo et al. confirmed that depression is associated with significantly increased levels of multiple inflammatory markers, including CRP, IL-6, and TNF-α. Notably, certain immune markers—such as CRP, IL-12, and sIL-2R—exhibit reduced variability in depressed patients, suggesting a more homogeneous inflammatory phenotype in this population [7]. Furthermore, approximately 20–30% of patients with depression fall into the category of “immuno-metabolic depression,” characterized by atypical energy-related symptoms, systemic low-grade inflammation, and metabolic abnormalities that include obesity, dyslipidemia, insulin and leptin resistance—features commonly associated with MASLD [8]. Nevertheless, the causal relationship between depression and MASLD remains controversial.
Although some studies have linked depression and MASLD through inflammation, the causal relationship is still controversial. Mendelian randomization (MR) studies, which use genetic instruments to mitigate confounding, have produced inconsistent results. For example, Lv et al. reported a significant causal effect of depression on MASLD risk [9], whereas Li et al. found no causal relationship [10].
The aforementioned evidence suggests that depression may promote the occurrence of MASLD through inflammation-centered immune dysregulation. To systematically validate this pathway, this study employs a multi-level integrated analytical strategy: first, cross-sectional epidemiological analysis based on National Health and Nutrition Examination Survey (NHANES) data is used to reveal the association between depression and MASLD and preliminarily screen the mediating effects of inflammatory and liver injury markers; second, two-sample Mendelian randomization analysis is applied to infer the causal direction between depression and MASLD and to validate the mediating effects of systemic inflammation (e.g., CRP) and liver enzymes (e.g., GGT, ALP) at the genetic level; next, transcriptomic co-expression network analysis combined with machine learning is conducted to identify shared immune-inflammatory molecular modules and hub genes linking depression and MASLD; finally, single-cell transcriptomic data are utilized to validate the dynamic expression of key molecules during disease progression at cellular resolution. This research framework, integrating macro-level associations, causal inference, and molecular mechanisms, aims to provide convergent evidence for the “depression–inflammation–MASLD” pathway and uncover the potential sex-specific immune mechanisms underlying it.
2. Materials and Methods
2.1. Study Design and Data Sources
A multilevel integrative design was employed, combining four complementary evidence streams: (i) a cross-sectional epidemiological analysis of depressive symptoms and MASLD using NHANES 2017–2020 data; (ii) a two-sample Mendelian randomization (MR) analysis to infer causality; (iii) transcriptome-wide co-expression network analysis combined with supervised machine learning to identify shared molecular mechanisms; and (iv) single-cell analyses to examine transcriptomic mechanisms at cellular resolution.
2.2. NHANES Disease Definitions and Analytic Strategy
NHANES 2017–2020 data were used for the cross-sectional analysis; the sample composition is shown in Table 1. MASLD was defined according to the 2023 international consensus, which requires evidence of hepatic steatosis—assessed by a controlled attenuation parameter (CAP) threshold—and at least one cardiometabolic risk factor: body-mass index (BMI) ≥ 25 kg/m^2^ or central obesity; impaired fasting glucose or hemoglobin A1c (HbA1c) ≥ 5.7% or diagnosed diabetes; elevated blood pressure or use of antihypertensive treatment; triglycerides ≥150 mg/dL or lipid-lowering therapy; or low high-density lipoprotein cholesterol (HDL-C). Participants with viral hepatitis or excess alcohol intake (>20 g/day in women; >30 g/day in men) were excluded. Depressive symptoms were assessed with the 9-item Patient Health Questionnaire (PHQ-9). The primary definition of depressive symptoms was PHQ-9 > 4; sensitivity analyses used thresholds of ≥10, ≥15, and ≥20, and PHQ-9 was also analyzed as a continuous measure [11].
Multivariable logistic regression models were constructed to examine the association between depression and MASLD. Models were estimated in the overall sample, adjusting for age and sex, followed by sex-stratified analyses. Effect modification by sex was formally evaluated using an interaction model including terms for depression, sex, their interaction, and age; the interaction coefficient and its 95% confidence interval (CI) were reported.
2.3. Mendelian Randomization
Two-sample MR was conducted to infer the causal relationship between depression and MASLD. Genetic instruments for depression were drawn from a meta-analysis by Howard et al., comprising 807,553 individuals of European ancestry (246,363 cases and 561,190 controls) [12]. Summary statistics for non-alcoholic fatty liver disease (NAFLD, used as a proxy for MASLD) were obtained from the FinnGen consortium (R10: 426,641 participants with 2568 cases; R12: 516,561 participants with 3649 cases) [13].
To evaluate mediation of the depression on MASLD’s effect by candidate biomarkers identified in the cross-sectional analysis, single-mediator and parallel-mediator analyses were performed using high-sensitivity C-reactive protein (hs-CRP) and liver enzymes (Alanine Aminotransferase [ALT], Aspartate Aminotransferase [AST], Gamma-Glutamyl Transferase [GGT], and Alkaline Phosphatase [ALP]) as candidate mediators. while a two-step MR mediation analysis was implemented. First, the causal effects of depression on circulating C-Reactive Protein (CRP), GGT, and ALP were estimated; second, the causal effects of CRP, GGT, and ALP on MASLD were estimated. Genome-Wide Association Study (GWAS) sources included the GWAS Catalog entries GCST90310151 [14], ukb-d-30730 [15], and GCST90468060 [16].
Instrument construction proceeded by clumping single-nucleotide polymorphisms (SNPs) associated with the exposure at genome-wide significance (p < 5 × 10^−8^); when instruments were sparse, the threshold was relaxed to p < 1 × 10^−6^. Linkage disequilibrium–based clumping used r^2^ < 0.001 within a 10,000 kb window to retain independent variants.
Indirect Effects were computed using the product-of-coefficients method. The total effect corresponded to the direct MR estimate for depression to MASLD. The significance of the mediation effect was assessed with the use of parametric bootstrap with 5000 replicate samples (increased to 10,000 if a near null distribution was required) to construct robust confidence intervals. Sensitivity analyses included: (1) triangulation across inverse-variance weighting (IVW), weighted median, weighted mode, simple mode, and MR-Egger; (2) tests for heterogeneity (Cochran’s Q), horizontal pleiotropy (Egger intercept), and outliers (MR-PRESSO); and (3) leave-one-out analyses to detect influential SNPs, together with Steiger filtering to confirm causal direction.
2.4. Transcriptome Data Analysis
An integrated bioinformatics and machine-learning framework was applied to identify genes shared between MASLD and depression. Two public datasets were retrieved from the Gene Expression Omnibus (GEO): GSE240729 (MASLD) [17] and GSE98793 (depression) [18]. Weighted gene co-expression network analysis (WGCNA) was performed separately on female samples from each dataset to construct disease-specific co-expression networks. Modules correlated with the phenotype were identified and subjected to Gene Ontology (GO) enrichment analysis. Modules with the strongest phenotype association and highest functional coherence were retained for integration.
To extract the most discriminative features, genes from depression-associated modules were entered into a supervised-learning pipeline. Using these module genes as predictors, multiple classifiers—support vector machine (SVM), random forest (RF), Lasso-penalized logistic regression, and XGBoost—were trained to classify depression status. Performance was evaluated by the area under the receiver-operating characteristic curve (AUC-ROC). The top 20 features by importance from the best-performing model were intersected with genes from the key MASLD module to define robust shared genes across the two disorders.
2.5. Single-Cell Analysis
Single-cell RNA-sequencing data from 45 liver samples (GEO: GSE202379) were integrated, including healthy controls (n = 6), NAFLD (n = 7), nonalcoholic steatohepatitis (NASH; n = 28), and NASH cirrhosis (n = 4) [19]. Given that CD4^+^ T cells are the principal immune subset expressing CD40LG and that its binding partner CD40 is predominantly expressed on B2 cells, lymphocytes and B2 cells were extracted for re-normalization, principal-component analysis, and clustering. CD4^+^ T cells were identified using a quantile-based dynamic threshold: a CD3 score was computed as the mean z-score of CD3D, CD3E, and CD3G expression; CD4 expression was taken from the normalized count matrix. Cells exceeding both the 70th percentile of the CD3 score and the 70th percentile of CD4 expression were classified as CD4^+^ T cells; remaining cells were labeled “other.” CD40LG expression in CD4^+^ T cells was compared across disease states, and CD40 expression was assessed in B2 cells.
2.6. Statistical Analysis
All analyses were conducted in R 4.4.3 and STRING. Key packages included TwoSampleMR 0.6.22 and MRPRESSO 1.0 for Mendelian randomization; WGCNA 1.73 for network analysis; caret 7.0-1, randomForest 4.7-1.2, e1071 1.7-16, xgboost 1.7.11.1, and lightgbm 4.6.0 for machine learning; and Seurat 5.3.0/SeuratObject 5.2.0 for single-cell analyses.
3. Results
3.1. Association Between Depression and MASLD in Population-Based Data
Clinical depression was significantly associated with a higher risk of MASLD in unadjusted models (Model 0: OR = 1.16, 95% CI [1.02–1.32]). After adjusting for age and sex (Model 1), the association remained significant (OR = 1.20, 95% CI [1.06–1.37]). Treating depression severity as a continuous variable (Model 2) indicated that each incremental increase in severity grade corresponded to a 14% higher likelihood of MASLD (OR = 1.14, 95% CI [1.06–1.23]). All variance inflation factors were below 10, suggesting no multicollinearity (Figure 1).
Sex-stratified analyses revealed significant effect modification by sex. Among females (Model 3), depression was strongly associated with MASLD risk (OR = 1.39, 95% CI [1.17–1.65]), whereas no significant association was observed among males (Model 4: OR = 0.99, 95% CI [0.81–1.20]). Comparable results were seen when using depression severity as the independent variable (Model 5: OR = 1.22, 95% CI [1.11–1.35]; Model 6: OR = 1.03, 95% CI [0.92–1.15]). An interaction test confirmed the sex-dependent pattern (p = 0.01). As illustrated in Figure 2a, in males, the predicted probability of MASLD was essentially unchanged regardless of depression status (0.70 vs. 0.69). In women, however, depression was associated with an increase in predicted MASLD probability, from 0.59 (95% CI [0.55–0.63]) to 0.68 (95% CI [0.64–0.72]) (Supplementary Table S1).
3.2. Inflammatory and Hepatic Injury Biomarkers Mediate Involved in the Association
We evaluated the potential mediating roles of hepatic enzymes (ALT, AST, GGT, and ALP) and the systemic inflammatory marker hs-CRP in the relationship between depression and MASLD. Multiple mediation analyses indicated that GGT, hs-CRP, and ALP exerted significant mediating effects. When these significant mediators were simultaneously included in a parallel mediation model, the association between depression and MASLD became fully mediated by hs-CRP (representing systemic inflammation) and by GGT and ALP (reflecting hepatic injury), rendering the direct effect nonsignificant.
3.3. Genetic Evidence for a Causal Effect of Depression on MASLD
To infer causality, we conducted two-sample MR analyses. Using the inverse variance–weighted (IVW) method, we observed a significant causal effect of genetically predicted depression on MASLD risk. This finding was consistently validated across two independent FinnGen cohorts—R10 (β = 0.483, SE = 0.146, p = 0.001) and R12 (β = 0.277, SE = 0.128, p = 0.031). The MR-Egger intercept test indicated no evidence of directional pleiotropy (p > 0.05), and MR-PRESSO analysis detected no outliers, supporting the robustness of the primary results (Figure 3a,b).
To explore potential mediation pathways between depression and MASLD, we used a two-step MR framework focusing on inflammation and liver injury markers. We separately evaluated systemic inflammation (CRP) and hepatic injury markers (GGT and ALP) as mediators. Two-step MR further confirmed that CRP (β_indirect = 0.008, p = 0.048), GGT (β_indirect = 0.095, p < 0.001), and ALP (β_indirect = 0.014, p = 0.010) jointly mediate the causal pathway, forming a genetic-level axis of depression → inflammation/liver injury → MASLD.
3.4. Transcriptomics Analysis Identified Immune-Inflammation Modules
Weighted gene co-expression network analysis (WGCNA) identified co-expression modules closely associated with immune and inflammatory responses in both depression and MASLD datasets—the turquoise module in depression and the cyan module in MASLD. (Figure 4a,b) Functional enrichment analysis revealed that genes in these top modules were significantly enriched for GO terms related to immune system processes (Figure 4c–h). KEGG pathway analysis revealed that the depression-associated turquoise module was enriched for B cell signaling and the NAFLD pathway, while the MASLD-associated cyan module involved T cell signaling (Figure 4i–l), collectively highlighting adaptive immunity as a potential shared mechanism.
Using feature genes derived from WGCNA, multiple machine learning models were constructed to identify potential biomarkers. The support vector machine (SVM) algorithm demonstrated the best predictive performance (AUC = 0.833) (Figure 4m). By intersecting the top 20 important genes from the SVM models of both conditions, we identified one common hub gene—CD40LG (Figure 4n).
3.5. Single-Cell Analysis: Upregulation of CD40LG in Intrahepatic T Cells During Disease Progression
A total of 5755 lymphocytes were extracted from the single-cell dataset, including 3600 cells from male samples and 2155 from female samples. Among them, 243 B2 cells were identified (142 male, 101 female). Using a quantile-based thresholding approach, screening thresholds were set at the 70th percentile for CD3 (score = 0.266) and CD4 (score = 0). Based on these criteria, 246 CD4^+^ T cells were identified, accounting for 4.3% of the total lymphocyte population.
The identified CD4^+^ T cells exhibited expected T cell marker gene expression patterns: canonical T cell markers were markedly elevated compared to other cells, including CD4 (14.5-fold), CD3E (4.0-fold), CD3D (3.3-fold), IL7R (2.2-fold), and CCR7 (3.7-fold). Expression of markers for other lymphocyte subtypes was very low in this population, confirming the specificity of the identification. These low markers included: CD8A (0.35-fold) and CD8B (0.26-fold) for CD8+ T cells; NKG7 (0.43-fold) and GNLY (0.47-fold) for natural killer (NK) cells; GZMB (0.27-fold), COBLL1 (0.64-fold), and TCF4 (0.67-fold) for plasmacytoid dendritic (pDC) cells; and TRGC1 (0.58-fold) and TRGC2 (0.50-fold) for γδ T cells (Figure 5a, Supplementary Tables S2 and S3).
Expression analysis showed that CD40LG expression in CD4^+^ T cells peaked at the NAFLD stage (mean = 0.452), then declined during NASH without cirrhosis (mean = 0.321), and remained relatively low and stable in late stages (NASH with cirrhosis and end-stage disease, mean ≈ 0.310). Similarly, CD40 expression in B2 cells peaked at NAFLD (mean = 0.008) before gradually declining. Due to small sample sizes, CD40 expression was undetectable in NASH with cirrhosis (n = 3) and remained lower in end-stage disease (mean = 0.04) than in earlier stages (Figure 5b).
Sex-stratified analyses further revealed distinct trends. In males, CD40LG and CD40 expression followed patterns similar to the overall cohort. In females, however, CD40LG expression in CD4^+^ T cells increased progressively with disease stage, while CD40 expression in B2 cells showed no consistent pattern, likely due to limited sample size (Figure 5c,d) (see Supplementary Tables S4 and S5).
4. Discussion
Our study integrates multi-level evidence from epidemiology, genetics, and transcriptomics to investigate the association between depression and MASLD. We established a causal link from depression to MASLD and identified immune-inflammatory pathways, as a key underlying mechanism, with a notably stronger effect in women.
Epidemiological analysis of NHANES data revealed a clear association between depression and increased MASLD risk, with a marked sex difference: depression was associated with elevated MASLD risk in women but not in men. Mediation analysis identified inflammatory and liver injury markers—hs-CRP (indirect effect = 0.048, p < 0.001), GGT (indirect effect = 0.054, p < 0.001), and ALP (indirect effect = 0.028, p < 0.001)—as complete mediators of the depression–MASLD relationship. These findings suggest that systemic inflammation may link depression to hepatic steatosis and metabolic dysfunction.
To clarify causal direction, we performed bidirectional two-sample MR. Genetically predicted depression was associated with increased MASLD risk (R10: OR = 1.621, 95% CI [1.217–2.160]; R12: OR = 1.319, 95% CI [1.026–1.697]), whereas reverse-direction analysis did not support an effect of MASLD on depression. Sensitivity analyses reinforced the robustness of these findings. Through mediated MR, we established a causal pathway from depression to MASLD via CRP, GGT, and ALP, providing genetic evidence for a unidirectional effect of depression on liver injury through inflammatory and metabolic mechanisms.
At the molecular level, WGCNA of female transcriptome samples showed that the top module in depression (turquoise) and a key module in MASLD (cyan) were both enriched for immune-related mechanisms, including immune regulation and cytokine signaling pathways. Machine learning approaches then identified CD40LG as a shared core immune regulatory molecule. CD40LG is a co-stimulatory molecule on T cells that can activate antigen-presenting cells (e.g., B cells, dendritic cells, and macrophages) and stimulate the release of inflammatory cytokines by binding to CD40 on these cells. The CD40LG–CD40 axis may act as an immune transducer, mechanistically linking depression-associated central immune activation to peripheral hepatic inflammation and metabolic dysfunction.
To further validate and elucidate the role of CD40LG, we analyzed its expression and that of its ligand CD40 across MASLD progression. In the overall sample, CD40LG in CD4^+^ T cells and CD40 in B2 cells exhibited stage-dependent activation, peaking at the MASLD stage. The upregulation of CD40LG in CD4^+^ T cells and CD40 in B2 cells suggests enhanced T cell–B cell interaction and an active adaptive immune response during the NAFLD stage. Similar patterns were observed in males. However, in females, CD40LG expression in CD4^+^ T cells increased progressively with disease stage, whereas CD40 expression in B2 cells showed no consistent trend, possibly due to limited sample size. The different trend of CD40LG between males and females may underlie the stronger depression–MASLD association in women. Overall, CD40LG has potential as an immunological biomarker for the inflammatory transition phase of NAFLD and as a target for future immunomodulatory interventions.
Given that CD40LG is located on the X chromosome, we consider the sex-specific regulatory mechanisms, such as escape from X-chromosome inactivation or hormonal modulation, could predispose females to stronger CD40LG-mediated immune signaling. In males, any CD40LG variant is hemizygous (only one X chromosome), while females can be homozygous or heterozygous. However, due to X chromosome inactivation (XCI) in somatic cells, typically only one X allele is active in females to balance expression. A study on CD40LG polymorphisms in Kawasaki disease analyzed eight loci and found only small, non-significant differences in allele frequencies between sexes [20], suggesting that for common variants the male–female difference in “CD40LG positivity” is likely minimal. Some literature indicates that escape from XCI may vary by cell type, but there is no consensus that CD40LG is a conventional XCI-escape gene [21]. We propose that sex differences in CD40LG expression or XCI escape may play a role, but this requires direct validation (e.g., allele-specific single-cell expression, RNA-FISH, or single-cell eQTL analyses).
Our findings suggest that CD40LG is not merely a marker of immune activation in MASLD, but may be involved in the critical transition from reversible metabolic impairment to irreversible inflammatory fibrosis, potentially mediating sex disparities. However, due to the limited number of cells, this conclusion merits further investigation in larger, sex-balanced cohorts. Future studies should validate CD40LG’s functional role in the liver microenvironment and explore its therapeutic potential as an immunomodulatory target, including its effects on sex-specific differences.
Furthermore, beyond genetic factors, sex hormones and neuroendocrine pathways may contribute to the observed sex disparity. Estrogen exhibits immunomodulatory properties that could influence inflammatory tone and adaptive immune responses, potentially altering CD40LG–CD40 signaling. Depression-associated dysregulation of the hypothalamic–pituitary–adrenal (HPA) axis and elevated cortisol may further promote hepatic inflammation and metabolic dysfunction [22]. These hormonal influences, together with X-linked genetic regulation, likely interact to shape the stronger depression–MASLD association observed in women.
The primary finding of our study—a unidirectional causal effect of depression on MASLD—is supported by several Mendelian randomization studies [9,23,24], strengthening the genetic evidence for depression as an upstream risk factor. The marked sex-specific association we observed, with effects concentrated in women, also aligns with epidemiological findings across diverse populations [9,25]. More importantly, our multi-omics approach provides mechanistic granularity: the female-predominant transcriptomic signature and the progressive upregulation of CD40LG in intrahepatic CD4^+^ T cells offer a plausible molecular substrate for this epidemiological and genetic signal. Although some prospective studies have suggested that baseline NAFLD may increase subsequent depression risk [26,27]—a finding not corroborated by our reverse MR analysis—the discrepancy between observational and genetic evidence may reflect methodological limitations and stage-dependent effects. In advanced disease stages, hepatic inflammation and psychosocial burden could jointly contribute to emotional disorders, forming a potential bidirectional cycle.
Our findings have important implications for clinical practice and public health. First, psychiatrists and hepatologists should be vigilant about comorbidity risks, especially in female patients with depression who also have metabolic syndrome or elevated inflammatory markers. It may be advisable to incorporate liver enzyme testing, hs-CRP measurement, and liver imaging into the routine management of these patients. Second, the blood biomarker CD40LG identified in this study holds promise for developing biomarker panels to identify high-risk individuals and enable targeted interventions. Third, our single-cell analysis suggests a potential intervention window early in MASLD progression. The peak expression of CD40LG in CD4^+^ T cells during the NAFLD stage indicates that immune activation via this axis is prominent during initial metabolic steatosis, before advanced fibrosis develops. This raises the possibility that early, depression-targeted anti-inflammatory strategies—whether pharmacological or behavioral—could attenuate immune-mediated hepatic injury and prevent transition to irreversible fibrotic disease.
This study has several limitations. First, the cross-sectional nature of the NHANES data restricts temporal inference, and the definitions of both depression and MASLD partly rely on subjective questionnaires, which may introduce bias. Despite multivariate adjustment, residual confounding such as diet and medication was difficult to completely rule out. Second, the observational mediation analysis depends on assumptions—such as correct temporal ordering and the absence of unmeasured confounding—that are difficult to fully verify. Moreover, the inflammatory marker hs_CRP used in the analysis is not specific to depression and may reflect processes related to other conditions or downstream effects rather than serving as a distinct causal intermediary. Third, in the mediation MR analysis, while we ruled out horizontal pleiotropy through methods such as MR-Egger and MR-PRESSO, the possibility of vertical pleiotropy cannot be completely excluded. Depression may elevate CRP, ALP, and GGT through other indirect pathways. Additionally, the transcriptomic data for depression were derived from peripheral blood, which may introduce tissue-specific noise. Finally, the functional mechanisms and clinical relevance of the CD40LG–CD40 axis, as well as the sex-specific patterns observed in the single-cell analysis, due to the limitation of single-cell data set, the stability of the results obtained is not strong, and further analysis is still needed in larger sample sizes and more balanced gender ratio data sets.
Building on our findings, several key research avenues emerge. First, prospective cohort studies measuring CD40LG (and related inflammatory markers) in depressed individuals are needed to validate its predictive value for MASLD development. Second, experimental models (e.g., chronic stress in hepatosteatosis models) should be used to mechanistically interrogate the role of the CD40LG-CD40 axis in linking neuroinflammation to liver pathology. Third, our study underscores the need for clinical trials to evaluate whether anti-inflammatory interventions or CD40 pathway modulation can improve metabolic outcomes in patients with comorbid depression and early-stage MASLD, particularly women. Finally, integrating metabolomic and lipidomic data in future multi-omics frameworks is crucial. Our genetic and transcriptomic evidence delineates a pathway from depression to MASLD; metabolomics can provide direct functional validation. For instance, NMR-based metabolomics reveals that depression is associated with a distinct serum profile—elevated triglycerides, VLDL, the inflammatory marker GlycA, and reduced glutamine—which is linked to MASLD-related metabolic dysfunction independently of liver disease severity [28]. This aligns with and functionally extends our inflammation-mediated hypothesis. Such integration could precisely map the downstream metabolic consequences of this immune-mediated pathway and identify clinically translatable biomarkers.
5. Conclusions
This study provides evidence that depression is a risk factor for MASLD, mediated substantially by inflammation and demonstrating a clear female preponderance. Furthermore, employing a multidisciplinary approach provides converging multi-omics evidence and generates a testable hypothesis regarding the CD40LG–CD40 axis. Collectively, our results argue for integrating liver surveillance into depression care and for exploring immunomodulatory strategies to reduce the prevalence of depression combined with MASLD, improve the quality of life of patients, and reduce medical costs.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1GBD 2021 Diseases and Injuries Collaborators. Global incidence, prevalence, years lived with disability (YL Ds), disability-adjusted life-years (DAL Ys), and healthy life expectancy (HALE) for 371 diseases and injuries in 204 countries and territories and 811 subnational locations, 1990–2021: A systematic analysis for the Global Burden of Disease Study 2021 Lancet 2024403213321613864257010.1016/S 0140-6736(24)00757-8PMC 11122111 · doi ↗ · pubmed ↗
- 2Miao L. Targher G. Byrne C.D. Cao Y.-Y. Zheng M.-H. Current status and future trends of the global burden of MASLD Trends Endocrinol. Metab. TEM 20243569770710.1016/j.tem.2024.02.00738429161 · doi ↗ · pubmed ↗
- 3Younossi Z.M. Kalligeros M. Henry L. Epidemiology of metabolic dysfunction-associated steatotic liver disease Clin. Mol. Hepatol.202431 S 3210.3350/cmh.2024.043139159948 PMC 11925440 · doi ↗ · pubmed ↗
- 4Shea S. Lionis C. Kite C. Lagojda L. Uthman O.A. Dallaway A. Atkinson L. Chaggar S.S. Randeva H.S. Kyrou I. Non-alcoholic fatty liver disease and coexisting depression, anxiety and/or stress in adults: A systematic review and meta-analysis Front. Endocrinol.202415135766410.3389/fendo.2024.1357664 PMC 1105898438689730 · doi ↗ · pubmed ↗
- 5Teixeira A.L. de Miranda A.S. Venna V.R. Himali J.J. Bauer M. E Understanding late-life depression: Focus on inflammation Curr. Opin. Psychiatry 20253837638210.1097/YCO.000000000000102240709634 · doi ↗ · pubmed ↗
- 6Kosekli M.A. Aktas G. The systemic immune inflammation index is a reliable and novel risk factor for metabolic dysfunction-associated fatty liver disease Curr. Med. Res. Opin.20254124725110.1080/03007995.2025.246395239912740 · doi ↗ · pubmed ↗
- 7Osimo E.F. Pillinger T. Rodriguez I.M. Khandaker G.M. Pariante C.M. Howes O.D. Inflammatory markers in depression: A meta-analysis of mean differences and variability in 5166 patients and 5083 controls Brain Behav. Immun.20208790190910.1016/j.bbi.2020.02.01032113908 PMC 7327519 · doi ↗ · pubmed ↗
- 8Penninx B.W.J.H. Lamers F. Jansen R. Berk M. Khandaker G.M. De Picker L. Milaneschi Y. Immuno-metabolic depression: From concept to implementation Lancet Reg. Health Eur.20254810116610.1016/j.lanepe.2024.10116639801616 PMC 11721223 · doi ↗ · pubmed ↗