From Orange to Oncology: Anti-Inflammatory and Anti-Cancer Mechanisms of Sinensetin
Dong Joon Kim, Songyeon Ahn, Xiaomeng Xie, Yeon-Sun Seong, Yong Weon Yi

TL;DR
Sinensetin, a compound found in citrus fruits, shows anti-inflammatory and anti-cancer effects by targeting multiple signaling pathways and immune responses.
Contribution
The paper provides a comprehensive review of sinensetin's mechanisms as a multi-target compound for cancer prevention and therapy.
Findings
Sinensetin suppresses inflammatory signaling and enhances antioxidant and autophagy programs.
It inhibits oncogenic pathways like β-catenin, PI3K/AKT, and VEGF in various cancer models.
Emerging evidence suggests sinensetin modulates immune cell activity and cytokine production.
Abstract
What are the main findings? Sinensetin is a citrus-derived flavone that consistently shows anti-inflammatory activities across models by suppressing inflammatory signaling and supporting antioxidant/autophagy programs.Across multiple cancers, sinensetin inhibits tumor-relevant signaling (including MKK6/P38 signaling), alongside emerging immune-related effects. Sinensetin is a citrus-derived flavone that consistently shows anti-inflammatory activities across models by suppressing inflammatory signaling and supporting antioxidant/autophagy programs. Across multiple cancers, sinensetin inhibits tumor-relevant signaling (including MKK6/P38 signaling), alongside emerging immune-related effects. What is the implication of the main finding? Multiple mechanisms support sinensetin as a multi-target lead for chemoprevention or adjunct therapy. Multiple mechanisms support sinensetin as a…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
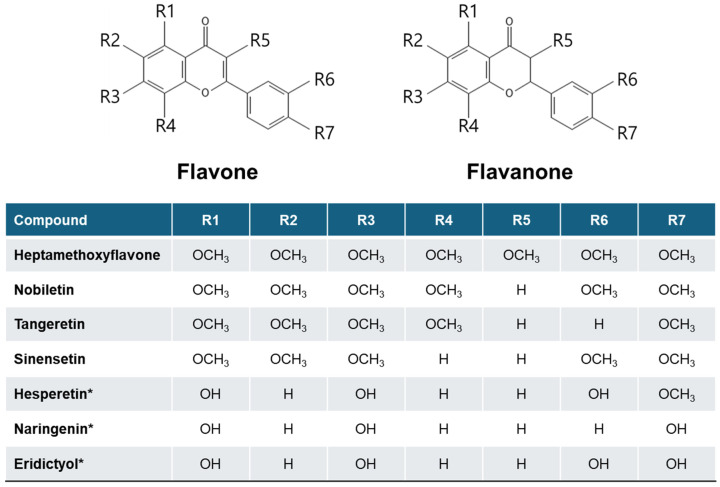 Figure 1
Figure 1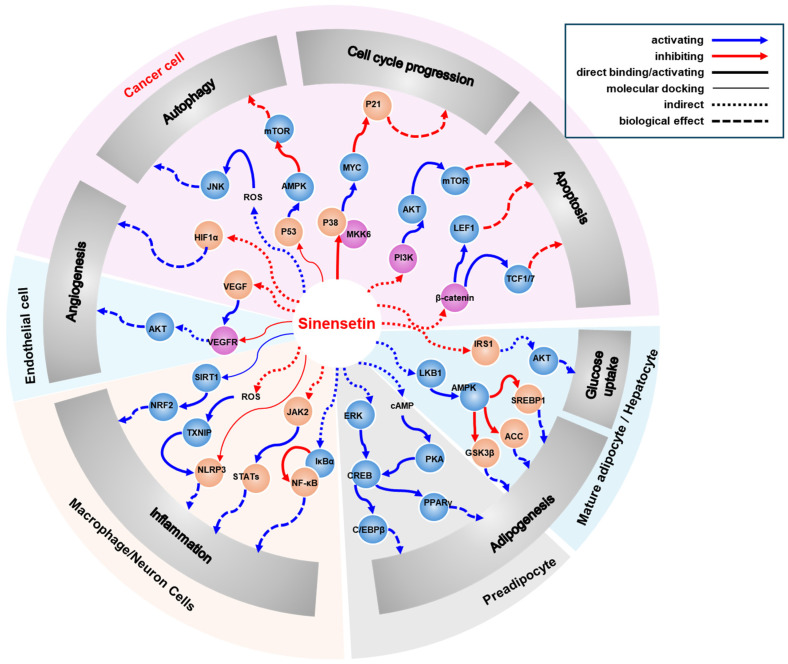 Figure 2
Figure 2- —Dankook University
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsBioactive Compounds in Plants · Tea Polyphenols and Effects · Microbial Metabolism and Applications
1. Introduction
Natural products have been explored as sources of dietary supplements and pharmaceuticals throughout human history due to their structural diversity and wide range of biological activities [1]. Orthosiphon aristatus (B1) Miq. [synonyms: O. stamineus Benth, O. grandifloras Bold., O. spicatus (Thumb.) Bak.; Lamiaceae] [2] has been widely used in traditional medicine across various countries, including China and parts of Europe, to treat conditions such as cancer, hypertension, rheumatism, tonsillitis, gout, menstrual disorders, diabetes, and diuresis [3,4,5,6,7]. One of the active compounds isolated from O. aristatus is sinensetin (Figure 1) [7,8,9]. Sinensetin is a pentamethoxy-flavone that is also present in several citrus fruits [10].
Generally, the most abundant flavonoids in sweet orange (Citrus sinensis) fruit and juice are flavanone glycosides such as hesperidin. Flavanones are known to be beneficial for human health, primarily through antioxidant activities. However, polymethoxyflavones (PMFs) have gained more attention due to the increasing evidence supporting their activities, including antioxidant, anti-inflammatory, anticancer, regulation of metabolic syndrome, and immune system, etc. [11]. Among several PMFs, Lam et al. showed that sinensetin exhibits the best anti-angiogenesis effect compared to nobiletin, hesperetin, scutellarein, neohesperidin, and naringin in HUVECs and zebrafish models [12]. Furthermore, sinensetin exhibits a broad spectrum of pharmacological activities, including anticancer, antioxidant, and anti-inflammatory effects [7,13,14]. Moreover, sinensetin has been shown to sensitize cancer cells to chemotherapeutic drugs by downregulating or inhibiting multidrug resistance proteins such as P-glycoprotein and ABCG2 (ATP-binding cassette subfamily G member 2), although the precise mechanisms underlying these effects remain unclear [8,15,16,17,18]. In this review, we examine the cellular signaling pathways, summarize in vitro and in vivo evidence for sinensetin’s anti-inflammatory, anticancer, and immune cell activities, and propose molecular targets with particular emphasis on our recent work identifying selective inhibition of MMK6 (mitogen-activated protein kinase kinase 6) [19]. Together, these mechanistic insights position sinensetin as a promising lead for chemopreventive and adjuvant therapeutic strategies.
2. Flavonoids in Oranges
Citrus fruits—such as oranges, mandarins, tangerines, grapefruits, and lemons—contain diverse flavonoids concentrated in the peel (flavedo/albedo) and, to a lesser extent, the pulp. These compounds act as antioxidants and modulators of inflammation, metabolic enzymes, and cell-signaling pathways [20]. In sweet orange (Citrus sinensis), the dominant flavanones in the juice and pulp are hesperidin, narirutin, and didymin, while the peel is enriched in PMFs such as nobiletin, tangeretin, and sinensetin (Table 1); other flavonoids (e.g., diosmin and anthocyanins in blood oranges) differ by the variety.
Hesperidin (hesperetin) activates the NRF2 (nuclear factor erythroid 2-related factor 2)—ARE (antioxidant responsive element) axis, upregulating phase-II/antioxidant defenses [e.g., NQO1 (NAD(P)H:quinone oxidoreductase 1), SOD (superoxide dismutase), and CAT (catalase)] and lowering reactive oxygen species (ROS). It also exerts anti-apoptotic and pro-survival effects through balancing the PI3K (phosphoinositide 3-kinase)/AKT and MAPK (mitogen-activated protein kinase) signaling. Together these mechanisms support anti-inflammatory and cytoprotective actions [25,26]. Narirutin (naringenin) shows anti-inflammatory and antioxidant activity via NF-κB (nuclear factor κB-light-chain-enhancer of activated B cells) downregulation and NRF2 upregulation, alongside metabolic benefits linked to AMPK (AMP-activated protein kinase) activation and suppression of lipogenesis [e.g., SREBP1c (sterol regulatory element-binding protein 1c)], contributing to improved lipid handling and insulin sensitivity in preclinical and emerging clinical data [27,28]. Didymin (diosmetin) inhibits the PI3K/AKT/mTOR (mammalian target of rapamycin) pathway and promotes caspase-dependent apoptosis in tumor models, with additional anti-inflammatory signals reported. These effects align with diosmetin’s pro-apoptotic and autophagy-inducing actions in cancer contexts [29,30].
Among PMFs, nobiletin improves metabolic profiles by activating AMPK and modulating lipid/glucose pathways (e.g., SREBP and lipogenic enzymes) and also enhances circadian clock output [e.g., CLOCK (circadian locomotor output cycles protein kaput)–BMAL1 (basic helix-loop-helix ARNT like 1)/ROR (receptor tyrosine kinase-like orphan receptor) axis], which is linked to anti-obesity and insulin-sensitizing effects in models [31,32]. Tangeretin attenuates the PI3K/AKT and ERK (extracellular signal-regulated kinase) signaling, induces cell-cycle arrest, and activates caspases-3/8/9, supporting antiproliferative and chemosensitizing effects in preclinical systems [33,34]. Lastly, sinensetin exhibits anti-inflammatory actions and inhibits angiogenesis by targeting various kinds of signaling, which we will discuss further in the following sections.
3. Anti-Inflammatory Mechanism of Sinensetin
A randomized, double-blind, placebo-controlled trial in adults with obesity found that the group that received flavonoid-enriched orange juice (treatment group) showed increased total antioxidant capacity concomitant with upregulation of GPX1 (glutathione peroxidase-1), indicating enhanced systemic redox defense [35]. Additionally, circulating pro-inflammatory cytokines decreased in the treatment group, including TNFα (tumor necrosis factor-alpha) and IFNγ (interferon-gamma), consistent with an anti-inflammatory effect of dietary PMF [35]. Together, this study suggests that citrus flavonoids can be effective clinically. However, there was no specific mechanism for the anti-inflammatory effect suggested in this study. There are several preclinical studies investigating the anti-inflammatory mechanisms of sinensetin (Table 2).
3.1. AMPK Pathway
The potential protective effect of sinensetin on osteoarthritis (OA) has been reported. Sinensetin did not exhibit cytotoxic effects on the primary chondrocytes at concentrations up to 40 μM, but it demonstrated anti-apoptotic effects in tert-butyl hydroperoxide (TBHP)-treated cells [36]. Additionally, sinensetin negatively regulated TBHP-induced extracellular matrix (ECM) degradation and promoted autophagy in TBHP-treated chondrocytes [36]. Similar to its effects in mature adipocytes [44], sinensetin increased phospho (p)-AMPK levels while decreasing p-mTOR levels in a dose-dependent manner [36].
Furthermore, AMPK-dependent regulation by sinensetin has been reported in a rat model of colitis. In 2,4,6-trinitrobenzene sulfonic acid (TNBS)-induced colitis model rats, sinensetin reduced inflammatory responses and enhanced autophagy of epithelial cells [37]. Sinensetin administration restored epithelial barrier functions by promoting autophagy-mediated inhibition of apoptosis and preventing claudin-2 degradation [37]. These protective effects on barrier functions were suggested to be mediated by the AMPK/ULK1 (Unc-51-like kinase 1) pathway, as siRNA-mediated depletion of AMPK abolished epithelial autophagy in Caco-2 cells [37].
3.2. NF-κB Pathway
Regulation of the NF-κB pathway by sinensetin has been reported in neuronal cells. Deposition of Aβ (amyloid β) peptides is considered to be a major pathological driver of Alzheimer’s disease (AD) [45]. Extracellular accumulation of Aβ aggregates induces neuronal toxicity through oxidative stress, inflammation, and apoptosis [45]. In cultured neuronal cells, sinensetin inhibited activation of the TLR4 (Toll-like receptor 4)/NF-κB pathway induced by Aβ_25–35_ [38]. Moreover, sinensetin dose-dependently reduced Aβ_25–35_-induced cytotoxicity in SH-SY5Y cells, but it did not exhibit cytotoxicity to SH-SY5Y cells in normal conditions [38]. Sinensetin also upregulated the expression of antioxidant molecules such as CAT, GSH (glutathione), and SOD in Aβ-treated cells [38]. In addition, sinensetin downregulated Aβ-induced inflammatory mediators, including nitrite, COX2 (cyclooxygenase-2; also known as PTGS2), IL1β (interleukin-1β), iNOS (inducible nitric oxide synthase; also known as NOS2), and TNFα [38]. Mechanistically, sinensetin attenuated the Aβ-induced upregulation of TLR4 and nuclear translocation of NF-κB P65 [38]. Since overexpression of TLR4 abrogated the effects of sinensetin on the Aβ-mediated neurotoxicity, apoptosis, and downregulation of antioxidant molecules, TLR4 is suggested as the major target for the neuroprotective effects of sinensetin [38]. Furthermore, sinensetin has been reported to inhibit BACE1 (β-site amyloid precursor protein-cleaving enzyme 1) activity in vitro [39], and BACE1 upregulation has been linked to NF-κB-mediated oxidative stress and inflammation in AD [46]. However, the precise mechanisms by which sinensetin regulates the TLR4/NF-κB axis remain to be elucidated.
In a lipopolysaccharide (LPS)-induced acute pulmonary inflammation mouse model, sinensetin attenuated lung injury by reversing the increases in CD68 (cluster of differentiation 68), MYD88 (myeloid differentiation primary response 88), and TLR4 expression through inhibition of the NF-κB pathway [40]. Molecular docking and molecular dynamics simulations suggested the potential binding of sinensetin to the NF-κB P65 subunit [47]. In LPS-treated mouse tissues, sinensetin reduced phosphorylation of IκBα (inhibitor of NF-κB) at S32/S36 residues, thereby enhancing the binding of IκBα to NF-κB P65 and downregulating NF-κB target gene expression, ultimately ameliorating LPS-induced acute pulmonary inflammation [40].
In human chondrocytes, while higher concentrations of sinensetin (≥5 μg/mL; ~13.4 μM) reduced cell viability, treatment with 1 μg/mL (~2.7 μM) exerted a protective effect against the IL1β-induced cytotoxicity [41]. Sinensetin decreased the secretion of inflammatory mediators such as COX2, IL6, iNOS, MMP13 (matrix metalloproteinase 13), and TNFα in IL1β-treated chondrocytes [41]. It also reversed the IL1β-mediated downregulation of collagen type II [41]. Transcriptome analysis and knockdown experiments identified SERPINA3 (serpin family A member 3) as a factor involved in the IL1β-mediated chondrotoxicity [41]. Sinensetin treatment upregulated SERPINA3 protein (alpha-1-antichymotrypsin) levels in IL1β-treated chondrocytes [41]. IL1β stimulation increased phosphorylation of IκBα and NF-κB P65, leading to nuclear localization of P65, whereas sinensetin suppressed phosphorylation of both proteins [41]. Consistently, sinensetin prevented cartilage degeneration in a rat OA model and reversed changes in SERPINA3, collagen II, and MMP13 expression [41]. Although the sinensetin-mediated upregulation of SERPINA3 is suggested as a mechanism for tis anti-inflammatory effects, no direct evidence has demonstrated that SERPINA3 inhibits NF-κB P65 nuclear translocation. Moreover, the mechanism by which sinensetin upregulates SERPINA3 remains unknown.
3.3. MAPK Pathway
In a cultured neurotoxicity model for oxygen–glucose deprivation/reperfusion (OGD/R) injury, sinensetin demonstrated anti-inflammatory and antioxidative effects though MAPK pathways. In human cerebral microvascular endothelial cells (HCMECs) subjected to OGD/R, sinensetin suppressed the OGD/R-induced upregulation of p-P38 (Y182), p-ERK1/2 (T185/T202), and p-JNK (JUN N-terminal kinase) (Y185) levels [42].
Interestingly, sinensetin reduced the IAV (influenza A virus)-induced upregulation of p-P38 in HEK293 cells without affecting the basal level of p-P38 in control HEK293 cells [43]. Recently, it has been reported that MKK6 is a direct target for sinensetin, modulating the MKK6/P38 pathway in non-small cell lung cancer (NSCLC) cells (will discuss later). Furthermore, sinensetin, along with nobiletin, was found to induce the transcriptional activation of CRE (cAMP response element) in the rat pheochromocytoma cell line PC12D [48]. Activation of the cAMP/PKA (protein kinase A)/ERK/CREB signaling by sinensetin has also been observed in cultured hippocampal neurons. Among the pharmacological components of Citrus reticulata, sinensetin was identified as the most potent activator of CRE-mediated transcription in rat hippocampal neurons in vitro [49]. Although the levels of p-CREB, p-ERK1/2, and p-PKA substrates were elevated by C. reticulata extracts, the specific effects of sinensetin on the individual signaling components have not yet been investigated.
4. Anticancer Mechanisms of Action
In addition to anti-inflammatory effects, it has been reported that sinensetin exerts anticancer effects through multiple pathways (Table 3).
4.1. β-Catenin Pathway
Anticancer effects of sinensetin through inhibition of the β-catenin pathway have been reported in breast cancer cells. Sinensetin reduced the viability of breast cancer cells (MCF7 and MDA-MB-231) by inducting apoptosis but did not affect the viability of normal mammary epithelial cells [50]. Sinensetin also suppressed invasion and epithelial–mesenchymal transition (EMT) of breast cancer cells, accompanied by the downregulation of β-catenin, LEF1 (lymphatic enhancing factor 1), TCF1 (T-cell factor 1)/TCF7 (transcription factor 7), and TCF3 (transcription factor 3)/TCF7L1 (transcription factor 7 like 1) at both the protein and mRNA levels [50]. Notably, treatment with a WNT (Wingless and Int-1) agonist reversed the sinensetin-mediated inhibition of cell survival, migration, EMT, and the expressions of β-catenin, LEF1, and TCF1/TCF7 proteins [50], suggesting that sinensetin may target WNT signaling or its downstream factors either directly or indirectly.
Regulation of the β-catenin pathway by sinensetin has also been reported in NSCLC cells and xenograft mouse models. Sinensetin inhibited the proliferation and migration of NSCLC cells in vitro and reduced the tumor size in xenograft mice [51]. In NSCLC cells, sinensetin suppressed EMT by downregulating p-AKT (T308), p-GSK3β (S9), and β-catenin protein levels [51]. Sinensetin also enhanced the cytotoxicity of CD8^+^ T-cells and increased the production of pro-inflammatory factors, such as IFNγ, IL2, and TNFα, thereby preventing immune evasion of NSCLC cells [51]. Importantly, treatment with the AKT activator SC79 [57] abrogated the sinensetin-mediated downregulation of p-AKT (T308), p-GSK3β (S9), and β-catenin both in vitro and in vivo, as well as the anti-tumor effects observed in vivo [51], indicating that AKT or its upstream regulators may be direct targets of sinensetin.
4.2. PI3K/AKT Pathway
The anticancer effects of sinensetin have also been reported in gallbladder cancer adenocarcinoma (GBAC). In the TJ-GBC2 GBAC cell line, sinensetin induced apoptosis through inhibition of the PI3K/AKT pathway [52]. Sinensetin reduced cell viability and migration in a dose-dependent manner and promoted apoptosis via downregulation of anti-apoptotic BCL2 (B-cell leukemia/lymphoma 2 protein) and upregulation of pro-apoptotic BAX (BCL2-associated X) expressions [52]. In addition, sinensetin inhibited cell migration and invasion by suppressing MMP2 (matrix metalloprotease 2) expression and enhancing MMP9 (matrix metalloprotease 9) expression [52]. Interestingly, sinensetin downregulated the levels of p-PI3K, p-AKT, and PTEN in GBAC cells [52].
Interestingly, potential binding of sinensetin to PI3K has been proposed based on molecular docking studies [58]. Furthermore, intragastric administration of sinensetin reduced the progression of pulmonary fibrosis (PF) in a bleomycin-induced PF mouse model [58]. Although sinensetin administration attenuated the bleomycin-induced upregulation of p-PI3K and p-AKT levels in pulmonary tissues [58], direct evidence of PI3K inhibition by sinensetin has not yet been demonstrated.
4.3. VEGF Pathway
The anti-angiogenic effects of sinensetin have been well-documented in a xenograft model with HepG2/C3A tumors. Sinensetin reduced the tumor volume and decreased the expression of neovascularization marker CD31, along with lower levels of VEGF (vascular endothelial growth factor) in tumor tissues [53]. Additionally, sinensetin inhibited both basal- and VEGF-induced proliferation, migration, and tube formation of human umbilical vein endothelial cells (HUVECs) in a dose-dependent manner [53]. Notably, sinensetin also diminished hypoxia-induced VEGF mRNA levels in HepG2/C3A, potentially through the downregulation of HIF1α (hypoxia-induced factor 1 α) [53]. Furthermore, sinensetin inhibited the VEGF-induced phosphorylation of VEGFR2 (VEGF receptor 2) and AKT in HUVECs [53]. Molecular docking analysis suggested that sinensetin might bind to the kinase active site of VEGFR2, thereby inhibiting the VEGF/VEGFR2/AKT signaling pathway [53]. However, further in vitro studies are necessary to confirm the binding and inhibition of VEGFR2 by sinensetin. Additional research is also needed to elucidate the mechanism underlying sinensetin-mediated downregulation of HIF1α in cancer cells.
Sinensetin’s effect on the VEGF/VEGFR2/AKT pathway has also been observed in lung adenocarcinoma (LUAD) cells. In these cells, sinensetin induced the expression of the microRNA (miR) miR-374c-5p, leading to the negative regulation of the VEGF/VEGFR2/AKT pathway [54]. The miR-374c-5p directly binds to the 3’-UTR (untranslated region) of VEGFA, resulting in decreased VEGFA expression [54]. Overexpression of VEGFA or inhibition of miR-374c-5p attenuated the anti-tumor effects mediated by sinensetin [54]. Moreover, sinensetin reduced the expression of cancer stem cell markers, including CD44 and CD133 [59], in LUAD cells [54]. In the context of NSCLC, the β-catenin/AKT pathway is implicated in immune evasion, and markers like CD44 and CD133 may play a role in this process [59]. However, the mechanism driving sinensetin-induced expression of miR-374c-5p remains to be elucidated and warrants further investigation.
4.4. Other Pathways
4.4.1. NRF2 Pathway
A virtual screening identified sinensetin as a potential NRF2 agonist [55]. In carbon tetrachloride (CCl_4_)-exposed HepG2 cells, sinensetin restored the expression of NRF2 and SOD1/2 and malondialdehyde (MDA) contents [55]. Furthermore, sinensetin increased the nuclear expression of NRF2 in CCl_4_-treated rats, which had reduced nuclear NRF2 levels compared to untreated control [55]. Although transient activation of NRF2, in normal cells in response to genotoxic stresses, has chemopreventive roles against carcinogenesis and tumor progression, persistent NRF2 activation may result in cancer progression, resistance to therapies, and poor prognosis [60]. Based on these backgrounds, further investigations are needed to determine the potential effects of sinensetin on the NRF2 pathway in the context of cancer.
4.4.2. P53 Pathway
Sinensetin has been shown to destabilize wild-type P53 in the hepatocellular carcinoma (HCC) cell line HepG2. Sinensetin induced autophagy in HepG2 cells while promoting apoptosis in Hep3B cells in a wild-type P53-dependent manner. In particular, sinensetin induced proteasome-dependent degradation of wild-type P53 in HepG2 cells [56]. It also reduced p-mTOR levels and increased p-AMPK and P27 levels [56]. Moreover, sinensetin promoted the nuclear translocation of P53 in HepG2 cells [56]. Blocking P53 translocation inhibited sinensetin-induced autophagy [56]. Interestingly, molecular docking simulations suggested that sinensetin could bind to the core domain of P53 [56]. The mechanism of sinensetin-induced autophagy is suggested to involve the P53/AMPK/mTOR pathway [56]. Contrary to its effects in HepG2 cells, sinensetin induced apoptosis in P53-null Hep3B cells [56]. Notably, sinensetin demonstrated minimal or no cytotoxic effects on normal human liver epithelial cells [56].
4.4.3. MKK6/P38 Pathway
A recent report [19] by our group suggested MKK6 as a putative target for sinensetin, using four independent techniques based on different principles: (1) in vitro kinase assay: Sinensetin selectively inhibited the phosphorylation of P38α by MKK6 but not MKK3. Purified proteins expressed in E. coli were used to exclude contamination from other mammalian kinases or substances. MKK6 kinase activity was measured by detecting p-P38 (T180/Y182) with a specific antibody; (2) in vitro binding assay: The direct binding of sinensetin to purified MKK6 was confirmed by surface plasmon resonance (SPR) analysis, showing an affinity of 66.27 μM in the presence of the substrate (ATP); (3) molecular docking: Although structural analysis of sinensetin-MKK6 complex was not experimentally proven, in silico molecular docking suggested that sinensetin binds to two separate helices (αC and αG) of MKK6. In contrast, sinensetin binds to only one helix (αC) of MKK3; and (4) siRNA-based knockdown experiment: Depletion of MKK6 by siRNA in NSCLC cells abrogated sinensetin-mediated cell growth inhibition. Sinensetin also demonstrated anticancer effects in NSCLC in an MKK6-dependent manner, potentially through induction of P21 expression and G1 phase cell-cycle arrest [19]. Unlike previous studies [61], the AKT or ERK pathway was not commonly regulated by sinensetin in NSCLC cells [19]. Based on molecular docking results, it has been suggested that sinensetin binds to the αG-helix of MKK6 to inhibit its substrate recognition [19]. However, further studies are needed to elucidate the precise molecular mechanisms for the differential binding of sinensetin to MKK6, given that the amino acid sequences of MKK6 and MKK3 have a complete match in their αG-helices and a high degree of similarity in the surrounding sequences [62].
5. Immunological Effects of Sinensetin
Tumor-associated macrophages (TAMs) have been implicated in tumor survival, metastasis, and immunity [63,64]. COX2 in TAMs has been reported to promote the survival and metastasis of breast cancer cells, and inhibition of COX2 prevents M2 macrophage polarization [64,65] and suppresses metastasis of breast cancer in a murine cancer model [66]. TAMs comprise M1-like and M2-like TAMs, and targeting M2-like TAMs has been recognized as a potential therapeutic target [67]. Substantial studies suggest potential role(s) of sinensetin in TAMs-mediated tumor progression and immunity (Table 4).
5.1. NF-kB Pathway
In macrophages, sinensetin appears to play a role in anti-inflammatory responses. Sinensetin reduced the expression of iNOS and COX2 in the murine macrophage cell line RAW264.7 following LPS treatment [68]. Consistently, LPS-induced mRNA levels for IL1β, IL6, and TNFα were downregulated by sinensetin in RAW264.7 cells [68]. Mechanistically, sinensetin delayed the degradation of IκBα, thereby inhibiting the nuclear translocation of the NF-κB P65 subunit [68].
As mentioned earlier, sinensetin attenuated LPS-induced acute pulmonary inflammation probably through binding to NF-κB P65 subunit [47] and reducing the levels of p-IκBα [40], leading to downregulation of NF-κB target gene expression.
5.2. SIRT Pathway
The anti-inflammatory effects of sinensetin have also been reported in an LPS-treated macrophage cell line model. Sinensetin attenuated the LPS-induced lung and liver injuries in mice, as evidenced by a reduction in serum ALT (alanine aminotransferase), IL1β, and TNFα [69]. In the murine macrophage RAW264.7 cells, sinensetin counteracted LPS-mediated modulation of various mRNAs. Specifically, sinensetin reduced LPS-induced upregulation of mRNA expressions for M1-type macrophage markers (iNos2, Cox2, and Cd86), while enhancing the LPS-suppressed mRNA expressions for the M2-type macrophage markers [Cd206, Cd68, and Agr1 (arginase 1)] [69]. Molecular docking analysis suggested that sinensetin binds to SIRT1 (sirtuin 1) [69]. Additionally, sinensetin mitigated the LPS-induced downregulation of Sirt1 and Nrf2 mRNAs in RAW264.7 cells [69]. Knockdown of Sirt1 attenuated the sinensetin-mediated NRF2 upregulation [69].
5.3. JAK2/STAT Pathway
Sinensetin also attenuated LPS-induced pulmonary inflammation in a mouse model. Along with the NF-κB pathway, sinensetin downregulated the levels of phosphorylated proteins in JAK2 (Janus kinase 2)/STAT pathways, including p-JAK2 (Y1008), p-STAT1 (S727), p-STAT3 (Y705), and p-STAT4 (Y693), in the LPS-treated mice and RAW264.7 macrophage cells [40].
Moreover, sinensetin reduced the expression of Nos2 mRNA and nitric oxide (NO) production in LPS-treated J774 cells (murine macrophage cells) in a dose-dependent manner [70]. It also reduced LPS-induced levels of COX2 and TNFα proteins and the production of prostaglandin E_2_ (PGE_2_). Interestingly, sinensetin reduced LPS-induced nuclear accumulation of STAT1α (signal transducer and activator of transcription 1α), with little or no effect on LPS-induced translocation of NF-κB P65 in J774 cells [70].
5.4. ROS/JNK Pathway
Sinensetin affects the apoptosis and autophagy in human T-cell lymphoma cells. In Jurkat cells, sinensetin induced autophagy by activation of JNK through accumulation of intracellular ROS [61]. The sinensetin-mediated induction of autophagy markers, such as the upregulation of LC3-II and Beclin-1 and downregulation of P62, were observed [61]. In addition, the activation of JNK was abrogated by treatment of N-acetyl cysteine (NAC), an ROS scavenger [61].
5.5. Interleukin (IL) Pathways
The immunomodulatory activity of sinensetin was studied in cyclophosphamide (CY)-induced immunosuppressed mice. In CY-treated mice, sinensetin suppressed CY-mediated body weight reduction and organ indices. In addition, sinensetin increased lymphocyte proliferation and secretion of IFNγ, IL2, and IL6 by upregulating the expression of mRNAs for Ifng, Il2, and Il6 in CY-treated mice [71].
The TXNIP (thioredoxin-interacting protein)/NLRP2 (NACHT, LRR, and PYD domains-containing protein 2)/Caspase 1/GSDMD (gasdermin D) pathway was also regulated by sinensetin treatment. In both the LPS-treated acute lung injury (ALI) mouse model and cultured RAW264.7 macrophage model, sinensetin attenuated inflammatory responses by inhibiting the dissociation of thioredoxin 1 (TRX1) and TXNIP and reducing the expression of ASC (apoptosis-associated speck-like protein containing a CARD), GSDMD, NLRP3, and pro-caspase 1 [72]. Mechanistically, sinensetin reduced the levels of ROS, and thereby inhibited the dissociation of TRX1 and TXNIP in ALI mice [72]. In addition, molecular docking analysis suggested a potential sinensetin-NLRP3 interaction [72].
6. Safety Aspects of Sinensetin
Although intensive investigations on the safety of sinensetin have not been performed yet [7], several studies showed its limited toxicity, especially on normal cells and tissues (Table 5). The maximum non-toxic dose of sinensetin is approximately 1.07 mM in human lung adenocarcinoma cell line A549 after 48 h treatment and assessed by cell counting kit-8 (CCK8) and lactate dehydrogenase (LDH) release assay [43]. However, a study by our group resulted in contradictory results in the antiproliferative effects on A549 cells when they were treated with 60 μM for 72 h in the 3-(4,5-dimethylthiazol-2-yl)-2-5-diphenyltetrazolium bromide (MTT) assay [19]. The maximum non-cytotoxic doses vary from 200 μM on normal human bronchial epithelial cells [51] and mouse primary T-cells [61] and approximately 2.69 μM on human chondrocytes [41]. Notably, a single-dose acute toxicity study in mice with 2000 mg/kg of sinensetin showed no significant toxicity and mortality for 2 weeks [55]. However, more intensive in vivo evaluations, such as toxicity, pharmacokinetic, pharmacodynamic, and biodistribution studies, are required before further clinical development.
7. Potential Targets for Sinensetin
For bioactive natural products, the identification of cellular targets (so-called target ID) is crucial for advancing new drug development [73,74,75,76,77]. Target ID is essential not only for understanding the mode of action (MoA) of a given natural product, but also for optimizing its specificity, selectivity, efficacy, and toxicity [73,74,75,76,77]. Although a plethora of technologies for target ID have been developed, the process remains challenging and often requires considerable time to yield validated targets [76]. As a result, many natural compounds still lack identified cellular targets [76].
Although the small molecular weight (372.4 g/mol) of sinensetin aligns well with Lipinski’s rule of five for drug-likeness [78] and it can be efficiently synthesized in high yield [79], its MoA remains elusive due to limited knowledge of its cellular target(s). Several enzymes have been reported as potential targets of sinensetin (Table 6). Molecular docking simulation is widely used to predict the potential interaction between small molecules and cellular target proteins [80]. Molecular docking analyses have suggested various cellular proteins, including AKT [81], AMPKγ [44], caspase 9 [47], COX2 [82], EGFR (epidermal growth factor receptor) [81,82], ERα (estrogen receptor α) [82], fibronetin [83], HIF1α [82], HSP90α (heat shock protein 90α) [81], MAPK3 [82], MYD88 [47], NF-κB P65 [47], NLRP3 [72], P53 [56,81], PI3K [58,81], PPARγ [82], SIRT1 [69], SRC [82], VEGFα [81], and VEGFR2 [53], as potential targets for sinensetin. Although some of these targets were linked to the biological effects of sinensetin, further studies are required to validate the interactions between sinensetin and its targets.
In vitro enzyme assays and spectral analyses, such as fluorescence quenching or circular dichroism (CD) spectra analysis with isolated proteins, are also widely used to screen or analyze small molecule–protein interactions. Using these techniques, several proteins have been identified as potential sinensetin targets, including α-amylase [5], BACE1 [39], BSA (bovine serum albumin) [85], CYP1A2 (cytochrome P450 1A2) [88], α-glucosidase [5,85], IFNγ [90], pancreatic lipase [91], and 15-lipoxygenae [92]. Although some of these were further supported by other in vitro assays or molecular docking simulations [39,85,89,90,91], other studies were based solely on in vitro enzyme assays [5,88,92]. One limitation of in vitro enzyme assays is their proneness to yield pseudo-positives. An example of this limitation is the artifact-prone fluorophore-based in vitro assay for SIRT1 activity, which led to a prolonged debate on the activation of SIRT1 by resveratrol [96,97]. Additionally, the biological consequences of sinensetin–target binding have not been validated in cellular contexts.
Cell-based assays are another method to identify cellular targets for given small molecules. As shown in Table 6, many proteins, such as CYP1A1/B1 [87], CYP3A4 [89], MATE1 (multidrug and toxin extrusion protein 1) [93], P-glycoprotein [18], SLCO1B1 (solute carrier organic anion transporter family member 1B1)/OATP1B1 [94,95], SLCO1B3/OATP1B3 [95], and SLCO2B1/OATP2B1 [95], have been identified as potential targets for sinensetin. Among them, only CYP3A4 was further studied with CD analysis and molecular docking [89]. One limitation of cell-based assays is their inability to differentiate direct effects from indirect or secondary effects. Although most cell-based assays use cells with overexpressed targets, it is difficult to completely exclude the indirect or secondary effects observed as assay readouts. Furthermore, cell-based assays are also susceptible to pseudo-results. For instance, luciferase-based assays sometimes result in false positive signals due to enzymatic interference with luciferase activity by a high proportion of compounds in libraries [98].
BACH1 (BTB and CNC homolog 1), a transcription regulator protein, is one of the most thoroughly studied potential targets for sinensetin. In a rat model, sinensetin ameliorated periodontitis in vivo [84]. BACH1 targeting by sinensetin was investigated using four techniques: (1) in silico molecular docking; (2) cellular thermal displacement assay; (3) knockdown; and (4) overexpression of BACH1. In silico molecular docking identified BACH1 as a sinensetin target [84]. The cellular thermal displacement assay further supported sinensetin-BACH1 binding, as the thermal stability of BACH1 increased in the presence of sinensetin [84]. Mechanistically, sinensetin binding to BACH1 reduced the levels of BACH1 via ubiquitin-dependent proteolysis and prevented its binding to the HMOX1 (heme oxygenase 1) promoter in periodontal ligament cells (PDLCs) [84]. However, the expression of HMOX1 protein was partially increased by sinensetin, exerting antioxidative responses in PDLCs [84]. Knockdown of BACH1 partially reduced sinensetin-induced HMOX1 expression [84]. Conversely, overexpression of BACH1 enhanced the effect of sinensetin on GSH contents [84]. Interestingly, BACH1 knockdown completely abolished the effects of sinensetin on SOD activity and MDA content in PDLCs [84]. Further studies are needed to investigate the mechanisms of BACH1 ubiquitination by sinensetin and its biological consequences.
As mentioned earlier, our group suggested MKK6 as a putative target for sinensetin [19]. An SPR analysis with purified MKK6 supported direct binding of sinensetin to MKK6 with an affinity of 66.27 μM in the presence of ATP. In vitro kinase assays further demonstrated the selective inhibition of MKK6 but not MKK3 by sinensetin. Molecular docking suggested that sinensetin binds to αC and αG helixes of MKK6, while it binds to only αC helix of MKK3. However, the molecular mechanisms for this selective inhibition of MKK6 remains to be determined.
8. Conclusions and Future Perspectives
Sinensetin, a citrus polymethoxylated flavone, has resurfaced as a multi-target natural product with promising anti-inflammatory and anticancer potential by affecting various cellular signaling pathways (Figure 2). In this review, we discussed that sinensetin modulates core inflammatory and oncogenic pathways—including AMPK, NF-κB, MAPK, β-catenin, AKT, P53, and MKK6—to suppress proliferation, migration, and survival signaling while enhancing antioxidant defenses and dampening cytokines such as TNF-α and IL-6. In neuronal and epithelial contexts, sinensetin restrains TLR4/NF-κB activation and supports barrier/autophagy programs via AMPK–ULK1, aligning with protective effects in colitis and OA models. Furthermore, sinensetin attenuated LPS-induced lung inflammation, down-modulated JAK2/STATs phosphorylations, and suppressed ROS-linked TXNIP/NLRP3 inflammasome activation in ALI models. In anticancer aspects, sinensetin inhibited β-catenin/LEF1/TCF signaling, reducing cell viability, EMT, and invasion in breast cancer. Suppression of AKT/GSK3β/β-catenin signaling impaired NSCLC growth. In GBAC cells, it restrained the PTEN/PI3K/AKT pathway, reduced migration, and promoted apoptosis. Beyond tumor cell-intrinsic effects, sinensetin exerts anti-angiogenic actions by lowering VEGF/HIF-1α, inhibiting VEGFR2 and AKT phosphorylations in endothelial cells, and reducing CD31^+^ neovascularization in LUAD xenografts. Immunologically, sinensetin enhanced anti-tumor immunity by boosting CD8^+^ T-cell cytotoxicity and increasing IFNγ/IL-2/TNFα in NSCLC models. Moreover, sinensetin modulated innate/adaptive responses in cyclophosphamide-immunosuppressed mice by improving lymphocyte proliferation and elevating IFNγ/IL-2/IL-6. However, most data are preclinical and often require mid- to high-micromolar concentrations. Therefore, future studies benchmarking other citrus PMFs to delineate structure–activity relationships are required, and this can lead to optimization for potency and selectivity. Closing these gaps will determine whether sinensetin advances from a promising multi-target natural product to a tractable candidate for precision anti-inflammatory, anticancer, and immunotherapy.
Decades-long efforts have been made to identify the cellular targets for sinensetin. As explored in this review, many cellular proteins have been suggested as potential targets for sinensetin. Among these, BACH1 and MKK6 have been intensively studied; however, targeting these proteins alone does not sufficiently explain all the reported biological effects of sinensetin. Additionally, the various biological activities of sinensetin have been observed in mid- and high-micromolar ranges, indicating a need to improve its potency through medicinal chemistry while reducing potential toxic adverse effects. Further validation of potential targets for sinensetin is crucial for advancing our understanding of its MoA and the future development of novel therapeutics.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Atanasov A.G. Zotchev S.B. Dirsch V.M. Orhan I.E. Banach M. Rollinger J.M. Barreca D. Weckwerth W. Bauer R. Bayer E.A. Natural Products in Drug Discovery: Advances and Opportunities Nat. Rev. Drug Discov.20212020021610.1038/s 41573-020-00114-z 33510482 PMC 7841765 · doi ↗ · pubmed ↗
- 2Awale S. Tezuka Y. Banskota A.H. Kadota S. Siphonols A–E: Novel Nitric Oxide Inhibitors from Orthosiphon stamineus of Indonesia Bioorg. Med. Chem. Lett.200313313510.1016/S 0960-894X(02)00854-512467611 · doi ↗ · pubmed ↗
- 3Beaux D. Fleurentin J. Mortier F. Effect of Extracts of Orthosiphon stamineus Benth, Hieracium pilosella L., Sambucus nigra L. and Arctostaphylos uva-ursi (L.) Spreng. in Rats Phytother. Res.19991322222510.1002/(SICI)1099-1573(199905)13:3<222::AID-PTR 447>3.0.CO;2-P 10353162 · doi ↗ · pubmed ↗
- 4Adam Y. Somchit M.N. Sulaiman M.R. Nasaruddin A.A. Zuraini A. Bustamam A.A. Zakaria Z.A. Diuretic Properties of Orthosiphon stamineus Benth J. Ethnopharmacol.200912415415810.1016/j.jep.2009.04.01419375494 · doi ↗ · pubmed ↗
- 5Mohamed E.A.H. Siddiqui M.J.A. Ang L.F. Sadikun A. Chan S.H. Tan S.C. Asmawi M.Z. Yam M.F. Potent α-Glucosidase and α-Amylase Inhibitory Activities of Standardized 50% Ethanolic Extracts and Sinensetin from Orthosiphon stamineus Benth as Anti-Diabetic Mechanism BMC Complement. Altern. Med.20121217610.1186/1472-6882-12-17623039079 PMC 3533584 · doi ↗ · pubmed ↗
- 6Mohamed E.A.H. Yam M.F. Ang L.F. Mohamed A.J. Asmawi M.Z. Antidiabetic Properties and Mechanism of Action of Orthosiphon stamineus Benth Bioactive Sub-Fraction in Streptozotocin-Induced Diabetic Rats J. Acupunct. Meridian Stud.20136314010.1016/j.jams.2013.01.00523433053 · doi ↗ · pubmed ↗
- 7Jie L.H. Jantan I. Yusoff S.D. Jalil J. Husain K. Sinensetin: An Insight on Its Pharmacological Activities, Mechanisms of Action and Toxicity Front. Pharmacol.20211155340410.3389/fphar.2020.55340433628166 PMC 7898666 · doi ↗ · pubmed ↗
- 8Mertens-Talcott S.U. Castro W.V.D. Manthey J.A. Derendorf H. Butterweck V. Polymethoxylated Flavones and Other Phenolic Derivates from Citrus in Their Inhibitory Effects on P-Glycoprotein-Mediated Transport of Talinolol in Caco-2 Cells J. Agric. Food Chem.2007552563256810.1021/jf 063138 v 17348674 · doi ↗ · pubmed ↗