Acacetin Attenuates Lysophosphatidylcholine-Induced Vascular Smooth Muscle Cell Injury via Sirt1-Nrf2/p62 Signaling Axis
Yun-Da Li, Yao Wu, Tian-Li Zhou, Qian Yuan, Gui-Rong Li, Wei-Yin Wu, Yan Wang, Gang Li

TL;DR
Acacetin protects vascular smooth muscle cells from injury by activating a specific signaling pathway, suggesting it could help stabilize atherosclerotic plaques.
Contribution
This study reveals a novel Sirt1-Nrf2/p62 signaling mechanism by which acacetin protects vascular smooth muscle cells.
Findings
Acacetin reduces LysoPC-induced apoptosis and ROS in vascular smooth muscle cells.
Nrf2 silencing eliminates the protective effects of acacetin, indicating its critical role.
In vivo experiments confirm acacetin's ability to suppress VSMC apoptosis and restore Sirt1 expression.
Abstract
Background: Acacetin, a naturally occurring flavone present in various plants, is known as a promising drug candidate for cardiovascular disorders. Our previous study demonstrated that acacetin ameliorates atherosclerosis through endothelial cell protection; however, its pharmacological effects on vascular smooth muscle cells (VSMCs) remain unexplored. This study investigates the therapeutic potential of acacetin against lysophosphatidylcholine (LysoPC)-induced VSMC injury and elucidates the underlying molecular mechanisms. Methods and Results: Multiple biochemical techniques were employed in the present study. The results showed that acacetin significantly attenuated LysoPC-induced apoptosis and reactive oxygen species (ROS) generation in cultured VSMCs. Western blot analysis revealed that the cytoprotection of acacetin was associated with upregulated expression of antioxidant defense…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
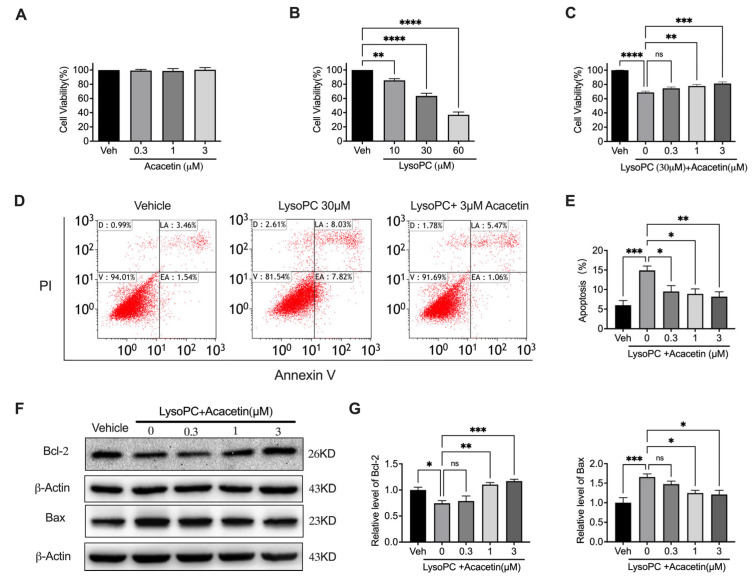 Figure 1
Figure 1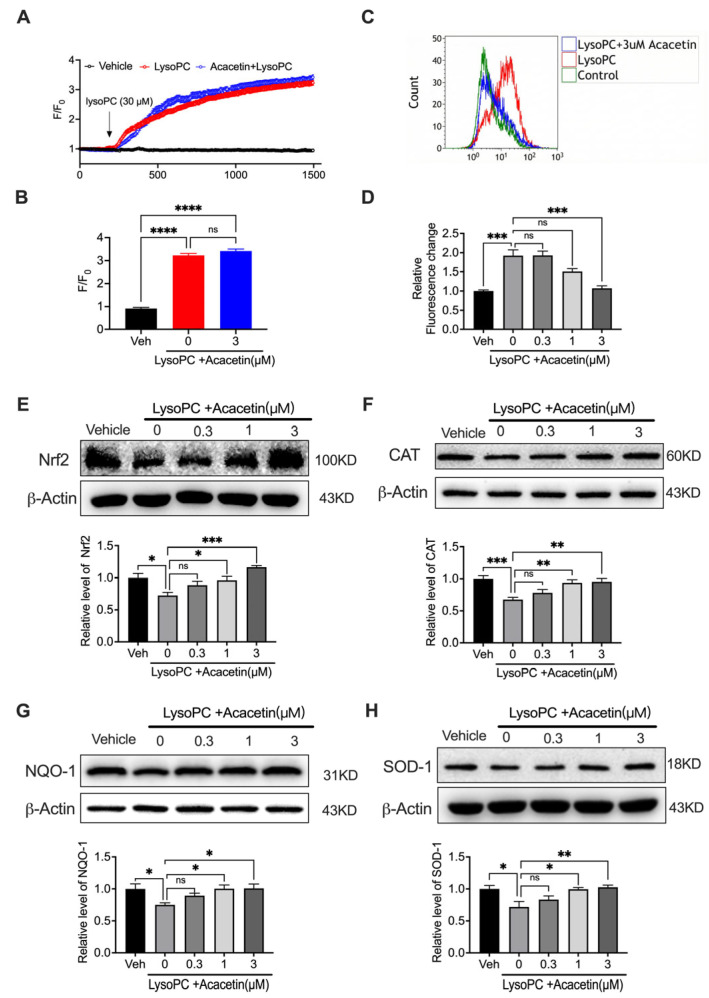 Figure 2
Figure 2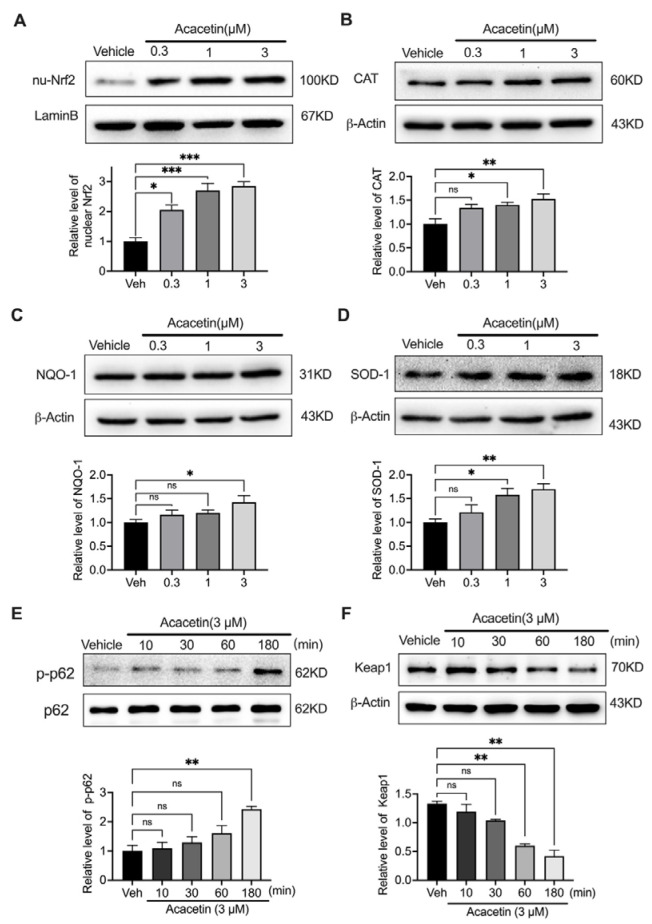 Figure 3
Figure 3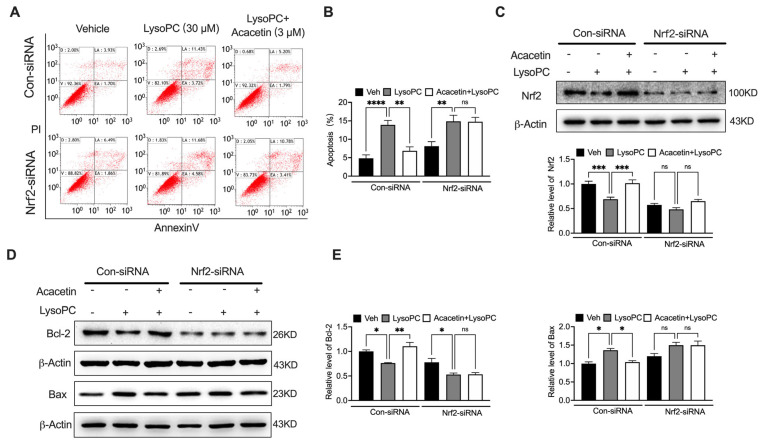 Figure 4
Figure 4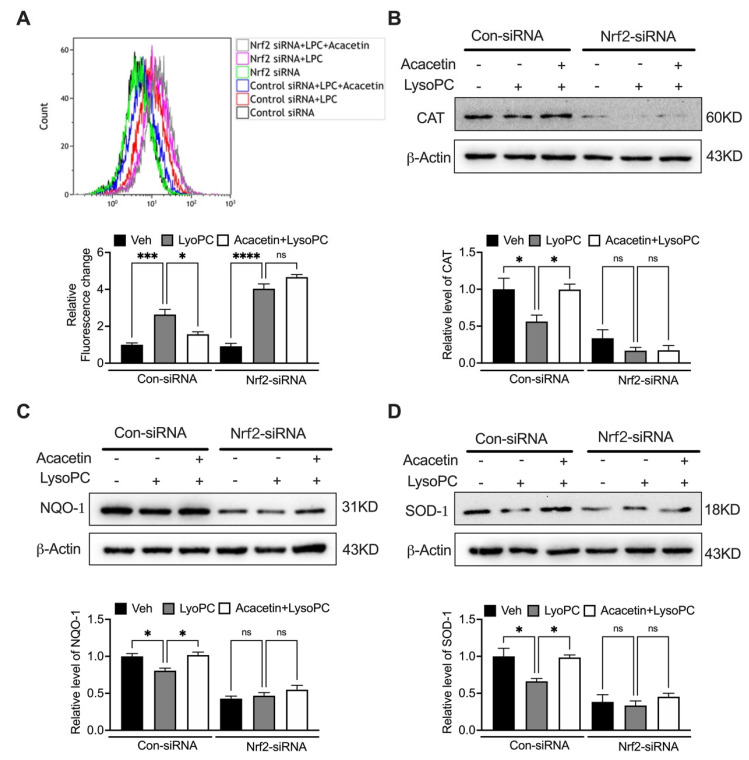 Figure 5
Figure 5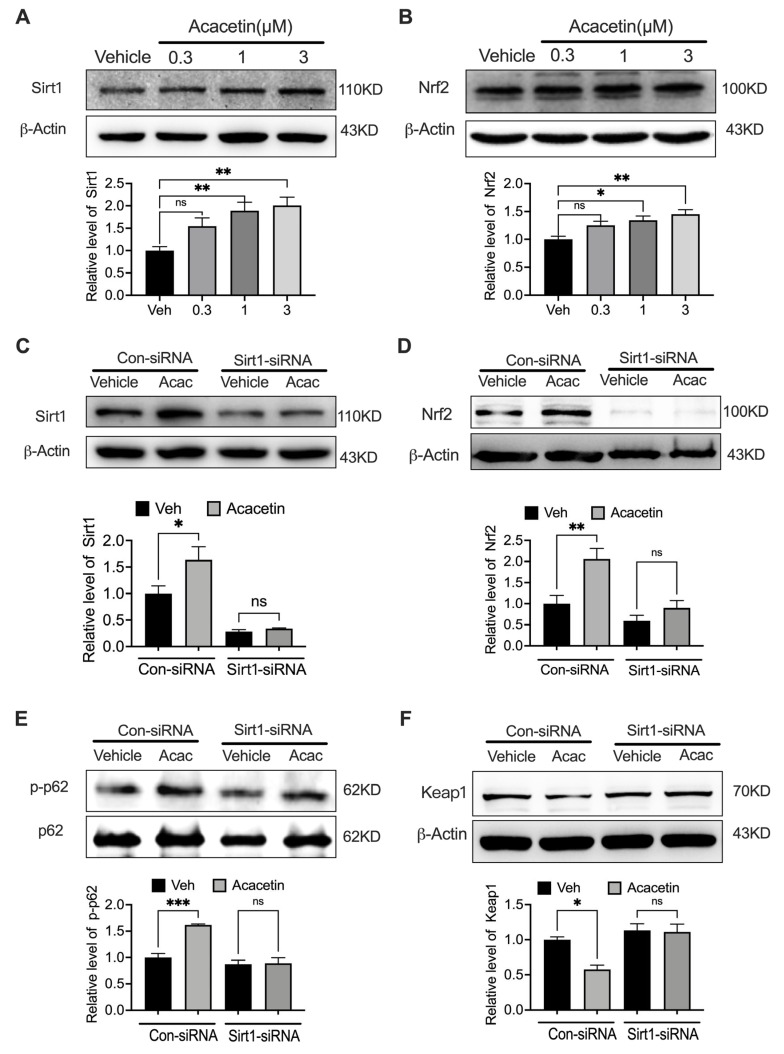 Figure 6
Figure 6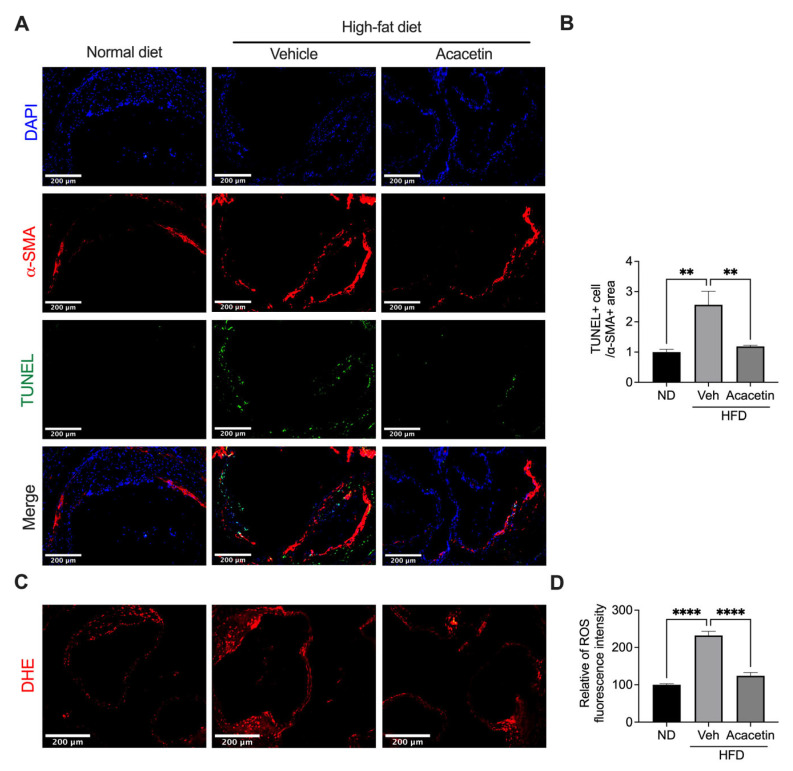 Figure 7
Figure 7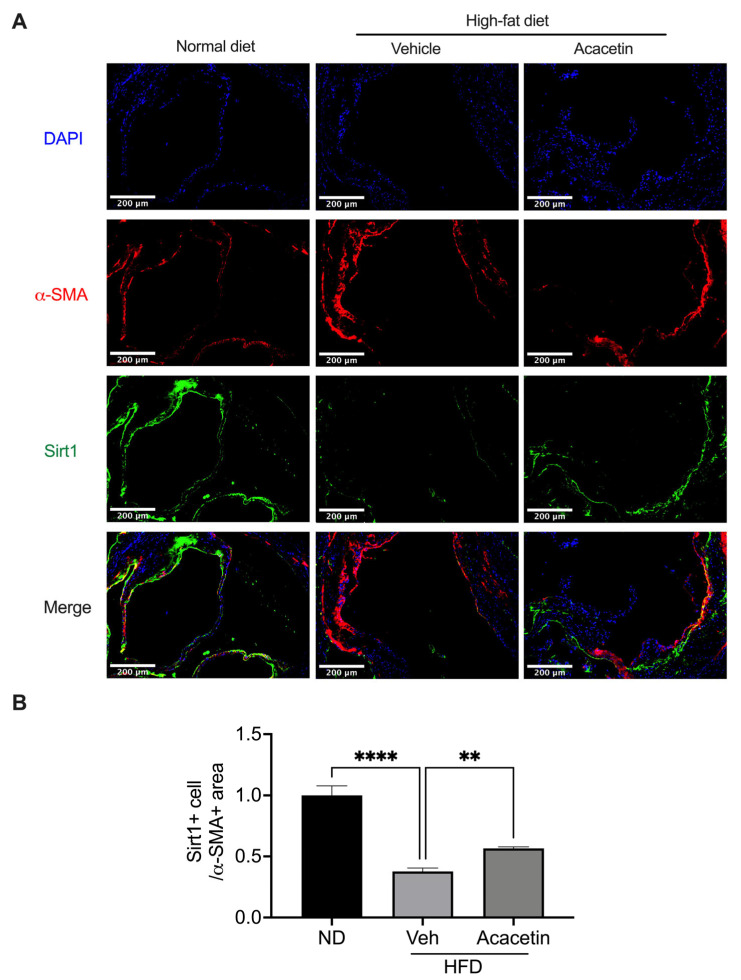 Figure 8
Figure 8- —National Natural Science Foundation of China
- —Fujian Provincial Health Technology Project
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsSirtuins and Resveratrol in Medicine · Medicinal Plants and Bioactive Compounds · Saffron Plant Research Studies
1. Introduction
Atherosclerosis (AS) is a vascular disorder characterized by dyslipidemia and chronic inflammation. Lysophosphatidylcholine (LysoPC), as the principal bioactive lipid component of oxidized low-density lipoprotein (ox-LDL), is significantly elevated in both plaque tissues and plasma of patients with atherosclerosis [1,2]. High plasma levels of LysoPC are positively correlated with plaque vulnerability and increased risk of adverse cardiovascular events [3]. LysoPC can directly act on VSMCs, inducing phenotypic switching, abnormal proliferation and migration, enhanced oxidative stress and inflammatory responses, and triggering various forms of programmed cell death [4,5,6]. These pathological alterations are closely associated with plaque instability, progression of vascular stenosis, and acute cardiovascular events in clinical settings [7,8].
Experimental evidence demonstrates that LysoPC may induce an elevation of cytosolic free Ca^2+^ in human coronary artery SMCs, rabbit coronary artery SMCs, and human umbilical vein endothelial cells [9,10,11]. Our prior mechanistic investigations further identified transient receptor potential canonical (TRPC) channels TRPC1 and TRPC3 as essential mediators of LysoPC-induced Ca^2+^ influx and subsequent apoptosis in human coronary artery SMCs. Genetic silencing of either TRPC isoform effectively abrogated LysoPC-triggered calcium overload and apoptotic signaling [12]. Despite these well-characterized pathogenic contributions, a critical translational gap persists: no currently available therapeutic agents specifically target LysoPC-mediated pathways to mitigate atherosclerotic progression. This unmet clinical need underscores the urgency for developing novel pharmacological strategies targeting LysoPC-associated molecular mechanisms.
Acacetin is a natural flavone compound ubiquitously present in Robinia pseudoacacia L., Pseudostellaria heterophylla, Chrysanthemum morifolium, and Pogostemon cablin [13,14] and exhibits a multifunctional pharmacological profile encompassing antimicrobial, anti-inflammatory, antioxidant, and antineoplastic properties [15,16,17,18,19,20], and diverse cardiovascular protective effects [14]. Beyond its well-documented atrial potassium channel blockade efficacy and atrial-selective atrial fibrillation [21,22,23,24], the cardio-protective potential of acacetin across diverse cardiovascular pathologies includes myocardial ischemia/reperfusion injury [25], cardiomyocyte hypoxia/reoxygenation damage [26], doxorubicin-induced cardiomyopathy and heart failure [27], pressure overload-induced cardiac hypertrophy [28], diabetic cardiomyopathy [29], and D-galactose-induced senescence-associated cardiomyopathy/heart failure [30]. Particularly relevant to vascular pathophysiology, acacetin demonstrates anti-atherosclerotic effects through endothelial protection mechanisms by activating Sirt1/Sirt3/AMPK signals under hyperglycemic conditions [31] and by activating the MsrA-Nrf2 pathway under Ox-LDL exposure [32]; however, the precise molecular mechanism behind the effects of acacetin on VSMCs, particularly under the specific stress with LysoPC, remains elusive. Although Nrf2 is the master regulator of antioxidant response, its canonical activation involves dissociation from Keap1. Recent evidence suggests a “non-canonical” pathway where the autophagy adaptor protein p62 (SQSTM1), upon phosphorylation, competes with Nrf2 for Keap1 binding. Whether acacetin is involved in the Sirt1/Nrf2/p62 axis to counteract LysoPC-induced vascular toxicity has not been investigated. To address this knowledge gap, the present study systematically evaluated the protective efficacy of acacetin against LysoPC-induced injury in rat aortic VSMCs and in aortic tissues of high-fat-diet-fed ApoE^−/−^ mice, employing a multidisciplinary experimental approach combining molecular biology and biochemical techniques.
2. Materials and Methods
2.1. Reagents and Antibodies
Acacetin (5,7-dihydroxy-4′-methoxyflavone) was chemically synthesized following established protocols with ≥99% purity [21] and water-soluble acacetin prodrug with 99.1% purity [22]. The in vitro concentrations 0.3–3 µM of acacetin and the in vivo dose 15 mg/kg/day (s.c.) of water-soluble acacetin prodrug used in this study were referred from the published literature [33].
The reagents including LysoPC and 2′,7′-dichlorofluorescein diacetate (DCFH-DA) were obtained from Sigma-Aldrich (St. Louis, MO, USA). Cell culture components comprising Dulbecco’s Modified Eagle Medium (DMEM), fetal bovine serum (FBS), 0.25% trypsin-EDTA, and antibiotic solutions (100 U/mL penicillin + 100 μg/mL streptomycin) were purchased from Thermo Fisher Scientific (Waltham, MA, USA), along with transfection reagents (Lipofectamine RNAiMAX, Opti-MEM) and calcium indicator Fluo-4 AM. Apoptosis detection was performed using an Annexin V-FITC/PI dual-staining kit from Dojindo Molecular Technologies (Kumamoto, Japan). Primary antibodies targeting Bcl-2 (ab32124), Bax (ab32503), Sirt1 (ab110304), Nrf2 (ab62352), Keap1 (ab119403), phospho-SQSTM1/p62 (Ser349, ab211324), catalase (CAT, ab16731), and SOD1 (ab13498) were procured from Abcam (Cambridge, UK), with additional antibodies against β-actin (sc-47778), Lamin B1 (sc-374015), NQO-1 (sc-32793), and p62/SQSTM1 (18420-1-AP) obtained from Santa Cruz Biotechnology (Dallas, TX, USA) and Proteintech (Rosemont, IL, USA), respectively. Gene silencing experiments utilized validated siRNAs targeting Nrf2 (sc-156128), Sirt1 (sc-108043), and non-targeting control (sc-37007) from Santa Cruz Biotechnology.
2.2. Cell Cultures
Primary vascular smooth muscle cells (VSMCs) were enzymatically isolated from thoracic aortic tissues of male Sprague-Dawley rats (200–250 g) following an established enzymatic digestion protocol [34]. Following isolation, cells were maintained in complete growth medium consisting of DMEM supplemented with 10% FBS, 100 U/mL penicillin, and 100 μg/mL streptomycin, under standardized culture conditions (37 °C, 5% CO_2_, 95% humidity). To ensure phenotypic stability and experimental consistency, all functional assays were conducted using cells between passages 3 and 8, corresponding to optimal viability and contractile marker preservation.
2.3. Cell Viability
VSMC viability was quantified using the MTT reduction assay according to standardized protocols [35]. Briefly, cells seeded in 96-well plates (5 × 10^3^ cells/well) were allowed to adhere for 12 h before pharmacological interventions. Cells were pretreated with pharmacologically relevant concentrations of acacetin (0.3, 1, and 3 μM)—dosages previously validated in cardiovascular models [26,27,28,32] or equivalent volumes of vehicle control (0.1% DMSO). Following 24 h co-incubation with LysoPC (30 μM) in low-serum DMEM (2% FBS), the MTT working solution (5 mg/mL in PBS, 100 μL/well) was introduced for 4 h at 37 °C. After careful aspiration of supernatants, formazan crystals were solubilized with anhydrous DMSO (150 μL/well) under gentle shaking. Optical density was measured at 570 nm using a microplate reader (BioTek Synergy H1, Agilent, Santa Clara, CA, USA), with viability values normalized to vehicle-treated control groups.
2.4. Aortic Tissue Acquisition and Experimental Design
The aortic root specimens analyzed in this study were derived from our established atherosclerosis model as previously reported [32]. Male ApoE^−/−^ mice (8-week-old, 22–25 g) were procured from Vital River Laboratory Animal Technology Co., Ltd. (Beijing, China) and maintained under specific pathogen-free conditions at Xiamen University Laboratory Animal Center (XMULAC). The experimental protocols were approved by the Ethic Committee for Animal Care and Use of Xiamen University (Approval No. XMULAC2020-0123). Following a 1-week acclimatization, mice were randomly stratified into three experimental cohorts: (1) Control group: Standard chow diet, (2) HFD group: Western diet (40% kcal fat, 1.25% cholesterol) for 12 weeks, and (3) HFD + Acacetin group: Western diet with concurrent subcutaneous acacetin administration (15 mg/kg/day) for 12 weeks. The therapeutic regimen was initiated synchronously with dietary induction to ensure temporal alignment of metabolic and pharmacological interventions. Acacetin was dissolved in vehicle solution (5% DMSO + 30% PEG300 + 65% saline) and administered via daily subcutaneous injection, while control groups received equivalent volumes of vehicle.
2.5. Cellular Apoptosis Quantification
Cellular apoptosis was assessed using dual-parameter flow cytometry with the Annexin V-FITC/PI Apoptosis Detection Kit (Dojindo, CK3001). For in vitro analysis, VSMCs seeded in 6-well plates (2 × 10^5^ cells/well) underwent pretreatment with acacetin (0.3, 1, 3 μM) or vehicle (0.1% DMSO in PBS) for 2 h prior to 24 h LysoPC (30 μM) challenge in 2% FBS DMEM. Following EDTA-free trypsinization (0.25% trypsin-EDTA, Thermo Fisher) and PBS (pH 7.4) washing, cells were re-suspended in 100 μL Annexin-binding buffer containing FITC-conjugated Annexin V (5 μL) and propidium iodide (5 μL) for 15 min dark incubation at RT. Flow cytometry analysis (Beckman Coulter Gallios, Brea, CA, USA) was performed within 1 h post-staining, with apoptotic populations quantified using Kaluza Analysis 2.1 software.
2.6. Plaque Apoptosis Analysis
In situ apoptosis within atherosclerotic plaques was evaluated through triple-i immunofluorescence-labeling using TUNEL/α-SMA/DAPI. Frozen aortic root sections (5 μm) underwent proteinase K digestion (20 μg/mL, 15 min) followed by TUNEL reaction mix (Beyotime, C1086) incubation (37 °C, 1 h). Sections were then blocked with 5% BSA and co-stained with α-SMA antibody (1:200, ab7817, Abcam) for 2 h at room temperature, followed by Alexa Fluor 594-conjugated secondary antibody (1:500, 1 h). Nuclear counterstaining with DAPI (1 μg/mL, 5 min) preceded mounting in anti-fade medium. Fluorescent images were acquired using the EVOS M7000 imaging system (Thermo Fisher) with consistent exposure settings across samples, and TUNEL^+^ α-SMA^+^ cells were quantified via ImageJ (v1.53) using threshold-based particle analysis.
2.7. Intracellular Calcium Flux Quantification
Dynamic intracellular free calcium ([Ca^2+^]i) fluctuations in VSMCs were monitored using real-time confocal imaging (Leica TCS SP5 II, Wetzlar, Germany) following established protocols [36,37]. Cells seeded in 35 mm-Dishes (Ibidi, 80136) at 2 × 10^4^ cells/cm^2^ density were serum-starved for 4 h prior to pretreatment with acacetin (0.3–3 μM) or vehicle (0.01% DMSO) in complete DMEM. Following pharmacological intervention, cells were loaded with 1 μM Fluo-4 AM (Thermo Fisher, F14201) in HEPES-buffered serum-free DMEM (pH 7.4) for 30 min at 37 °C under 5% CO_2_ humidified atmosphere. Post-incubation, three sequential washes with pre-warmed (37 °C) Tyrode’s solution (in mM: 140 NaCl, 5 KCl, 1 MgCl_2_, 2 CaCl_2_, 10 HEPES, 10 glucose, pH 7.4) were performed to remove extracellular dye. Time-lapse imaging was conducted at 23–25 °C using 488 nm excitation/515–530 nm emission settings, with images acquired at 2 s intervals over 300 s. Fluorescence intensity quantification was performed using Leica LAS X software (v3.7.4), with data normalized to baseline F_0_ values.
2.8. Reactive Oxygen Species (ROS) Quantification
Cellular ROS generation was assessed using DCFH-DA following established protocols [26]. VSMCs seeded in 6-well plates (1 × 10^6^ cells/well) were pretreated with acacetin (0.3, 1, 3 μM) or vehicle (0.05% DMSO) for 2 h prior to LysoPC (30 μM) stimulation in 2% FBS DMEM. For siRNA-mediated gene silencing experiments, cells transfected with Nrf2-targeting siRNA (50 nM) or scramble control (Santa Cruz, sc-37007) using Lipofectamine RNAiMAX were cultured for 48 h for protein knockdown before 24 h acacetin (3 μM) treatment. Following treatments, cells were loaded with 10 μM DCFH-DA in serum-free DMEM (37 °C, 30 min, dark), washed twice with Hanks’ Balanced Salt Solution (HBSS), and immediately analyzed by flow cytometry (Beckman Coulter Gallios, excitation/emission: 488/525 nm). A minimum of 10,000 events per sample were acquired, with fluorescence intensity quantified using Kaluza Analysis 2.1 (Beckman Coulter).
2.9. Vascular Oxidative Stress Assessment
In situ ROS production in atherosclerotic plaques was evaluated through dihydroethidium (DHE) fluorescence imaging. Aortic roots embedded in optimal cutting temperature (OCT) compound (Sakura, 4583, Tokyo, Japan) were cryosectioned into 5 μm thick slices using a CM1950 cryostat (Leica, Wetzlar, Germany). Sections were equilibrated to RT, incubated with 10 μM DHE (Sigma-Aldrich, D7008) in PBS (37 °C, 20 min, dark), and counterstained with DAPI (1 μg/mL, 5 min). After three PBS washes (5 min each), sections were mounted with ProLong Diamond Antifade (Thermo Fisher, P36961) and imaged under consistent parameters (40× objective, 561 nm excitation/610 nm emission) using the EVOS M7000 imaging system. DHE fluorescence intensity (≥5 fields/sample) was quantified using Image J (v1.53) with standardized threshold settings.
2.10. Nucleoprotein Extraction
The nuclear protein of Nrf2 was isolated using a Nuclear and Cytoplasmic Protein Extraction Kit (Beyotime, Beijing, China) in accordance with the manufacturer’s instructions. Unless stated, all steps were performed at 4 °C or on ice.
2.11. siRNA Transfection
Transfection of cells with siRNA was performed using Lipofectamine RNAiMAX transfection reagent, according to the manufacturer’s instructions. VSMCs at 60~70% confluence were transfected with siRNA molecules targeting rat Nrf2, Sirt1, and negative control at 100 nM. After transfection for 48 h, cells were used for determining ROS generation, protein expression, and cell apoptosis assays.
2.12. Western Blot Analysis
Western blot was performed as described previously [36]. Briefly, cells were lysed with a modified RIPA buffer (Solarbio, Beijing, China) containing 1% protease and phosphatase inhibitors (Roche, Germany). The protein concentration was determined using the BCA Protein Assay Kit (Thermo Fisher Scientific, Carlsbad, CA, USA). Cell lysates were mixed with sample buffer (5× loading buffer) and denatured by heating to 95 °C for 5 min. An equal amount of protein for each sample was resolved via 10% or 12% SDS-PAGE gel and then transferred to immunoblot PVDF membranes (Bio-Rad, Hercules, CA, USA). Membranes were blocked with 5% non-fat milk in Tris buffer saline with 0.1% tween-20 (TBST) for 1h, followed by overnight incubation at 4 °C with specific primary antibodies (at a range from 1:1000 to 1:2000). After being washed with TBST three times, the membranes were incubated with HRP-conjugated secondary antibodies at 1:10000 dilutions in TBST at room temperature for 1 h. Membranes were rewashed with TBST and then visualized with ECL reagents (Advansta, Menlo Park, CA, USA). The signals were detected with a chemiluminescence detection system (FluoChem E, Biotechne, Minneapolis, MN, USA). The relative band intensities were quantified by Image J software (NIH, Bethesda, MD, USA).
2.13. Statistical Analysis
Statistical analysis was performed using GraphPad Prism 9 (GraphPad Software, Inc., San Diego, CA, USA). Results are presented as means ± SEM. One-way ANOVA followed by post hoc Tukey’s test was used for comparison among groups. A value of p < 0.05 was considered statistically significant.
3. Results
3.1. Acacetin Mitigates LysoPC-Induced VSMC Apoptosis Through Bcl-2/Bax Regulation
As shown in Figure 1, acacetin showed significant cytoprotective effects against LysoPC-induced apoptosis in rat aortic VSMCs. Consistent with previous findings in human umbilical vein endothelial cells (HUVECs) [31], acacetin (0.3–3 μM) exhibited no intrinsic cytotoxicity in VSMCs across all tested concentrations (Figure 1A, n = 6). LysoPC treatment (10–60 μM) concentration-dependently reduced cellular viability (Figure 1B, n = 8). Pharmacological intervention with acacetin moderately ameliorated LysoPC (30 μM)-induced viability loss (Figure 1C, n = 9). Flow cytometric quantification revealed that LysoPC triggered an increase in apoptotic population, which was substantially attenuated by acacetin pretreatment (Figure 1D,E, n = 8). Mechanistically, Western blot analysis demonstrated that LysoPC exposure (30 μM, 24 h) significantly downregulated the anti-apoptotic protein Bcl-2 while upregulating pro-apoptotic Bax (Figure 1F,G, n = 5). Acacetin reversed these perturbations in a concentration-dependent manner. These findings show that acacetin confers cytoprotection against LysoPC-induced VSMC apoptosis through increasing Bcl-2 protein expression and decreasing Bax protein expression, suggesting its potential therapeutic utility in stabilizing atherosclerotic plaques through VSMC preservation.
3.2. Acacetin Suppresses LysoPC-Induced ROS Over-Production
Building upon our prior demonstration of TRPC1/3-mediated Ca^2+^ dysregulation in LysoPC-induced human coronary SMC apoptosis [12], we investigated whether acacetin’s anti-apoptotic effects involved calcium modulation. Confocal imaging revealed 30 μM LysoPC triggered an increase in cytosolic Ca^2+^, which persisted despite 3 μM acacetin co-treatment (Figure 2A,B, n = 4), effectively excluding Ca^2+^-dependent mechanisms in acacetin’s cytoprotection.
We subsequently investigated whether ROS-mediated signal pathway involved the cytoprotection of acacetin using DCFH-DA flow cytometry. LysoPC (30 μM) induced a ROS surge, which was concentration-dependently mitigated by acacetin (Figure 2C,D, n = 4). The ROS attenuation paralleled the anti-apoptotic efficacy of acacetin, suggesting that redox regulation is the primary protective mechanism. Mechanistic investigation of the Nrf2 antioxidant pathway demonstrated LysoPC significantly suppressed key mediators: Nrf2, catalase, NQO1, and SOD1. Acacetin (0.3–3 μM) concentration-dependently restored their expression (Figure 2E–H, n = 4–6). Strikingly, acacetin at 3 μM normalized Nrf2 nuclear translocation, confirming pathway activation. These data establish that acacetin counteracts LysoPC-induced VSMC apoptosis primarily through Nrf2-driven transcriptional reactivation of antioxidant response, effectively neutralizing oxidative stress independent of Ca^2+^ signaling pathways.
3.3. Acacetin Orchestrates Nrf2 Nuclear Translocation via p62/Keap1 Axis
The Nrf2-Keap1-p62 regulatory triad constitutes a pivotal oxidative stress response system [38]. Under basal conditions, Nrf2 forms a complex with Keap1 in the cytoplasm, while p62 phosphorylation at Ser349 facilitates competitive Keap1 binding, enabling Nrf2 liberation and nuclear translocation to activate antioxidant genes [39,40]. Western blot analysis revealed acacetin concentration-dependently enhanced nuclear Nrf2 accumulation (Figure 3A, n = 4), concomitant with upregulated expression of downstream effectors CAT, NQO1, and SOD1 (Figure 3B–D, n = 5–6). Temporal analysis demonstrated that 3 μM acacetin progressively increased p62 phosphorylation (Ser349) while reducing Keap1 levels (Figure 3E,F, n = 4). These findings present a novel mechanism whereby acacetin promotes Nrf2 nuclear shuttling through coordinated p62 activation and Keap1 suppression.
3.4. Genetic Ablation of Nrf2 Nullifies Cytoprotective Effects of Acacetin
SiRNA Silence of Nrf2 completely abolished acacetin-mediated protection. In scramble controls, acacetin (3 μM) reversed LysoPC-induced viability reduction and apoptosis, while Nrf2-silenced cells remained unresponsive (Figure 4A–C, n = 5–8). Similarly, the modulation of Bcl-2 increase and Bax decrease by acacetin was abrogated upon Nrf2 depletion (Figure 4D,E, n = 4–6). Consistent with apoptotic regulation, the antioxidant capacity of acacetin proved to be Nrf2-dependent. While acacetin attenuated LysoPC-induced ROS surge in control cells, this effect was absent in Nrf2-deficient VSMCs (Figure 5A, n = 4). The rescue of antioxidant enzymes by acacetin—CAT, NQO1, SOD1—strictly required Nrf2 expression (Figure 5B–D, n = 4–5). These data unequivocally position Nrf2 as the central mediator of acacetin’s pleiotropic protective effects.
3.5. Sirt1 Governs Acacetin-Induced Nrf2 Activation in VSMCs
Building on our prior demonstrations of Sirt1-dependent Nrf2 regulation in cardiovascular cells [27,28,30,31], we identified a conserved mechanism in VSMCs. Acacetin concentration-dependently upregulated Sirt1 and Nrf2 protein levels (Figure 6A,B, n = 5–6). Sirt1 silencing prevented acacetin-induced: Sirt1/Nrf2 protein elevation (Figure 6C,D, n = 4–5), p62 phosphorylation (Ser349, Figure 6E, n = 4), and Keap1 downregulation (Figure 6F, n = 4). This comprehensive analysis establishes Sirt1 as the upstream regulator of acacetin’s antioxidant signaling through the Nrf2/p62/Keap1 axis, revealing therapeutic targeting potential of acacetin in vascular pathologies.
3.6. Acacetin Rescues Sirt1-Mediated Vascular Homeostasis in Atherosclerotic ApoE−/− Mice
Building upon our prior demonstration of acacetin’s potent anti-atherogenic efficacy in HFD-fed ApoE^−/−^ mice [32], we herein demonstrate its cellular protective mechanisms within aortic tissues. Histomorphometric analysis revealed HFD-induced profound VSMC apoptosis and oxidative stress, which were markedly attenuated by chronic acacetin treatment (15 mg/kg/day, 12 weeks, Figure 7A–D, n = 7–9). This in vivo protection mirrored our in vitro findings with LysoPC-challenged VSMCs, validating the pathophysiological relevance of our mechanistic model. Mechanistically, immunoblot quantification demonstrated HFD-driven Sirt1 downregulation in aortic lysates, which was significantly rescued by acacetin (Figure 8A,B, n = 7). These translational findings substantiate that acacetin’s vascular protective effects in complex atherosclerotic milieus operate through conserved Sirt1 activation mechanisms, bridging cellular-level discoveries with whole-organism pathophysiology.
4. Discussion
LysoPC induced VSMC apoptosis and inflammation mediated by concurrent calcium dysregulation and ROS overproduction [12], while the results from the present study support the notion that apoptosis and inflammation induced by LysoPC are effectively countered by acacetin via restoring the damaged redox homeostasis rather than calcium homeostasis modulation. This study elucidates a vasoprotective signal axis of acacetin targeting Sirt1/Nrf2/p62 pathway in VSMCs. Acacetin orchestrates antioxidant response through sequential Sirt1 activation → Nrf2 nuclear translocation → transcriptional upregulation of NQO-1/SOD1, establishing a multi-tiered protective cascade in VSMCs.
Our previous studies have demonstrated that acacetin has significant cardiovascular protection in different in vitro and in vivo models, in which the Sirt1-mediated signaling cascade is involved [14]. In addition to reversing hypoxia pulmonary hypertension via inhibiting pulmonary artery VSMC hyper-proliferation via Sirt1-HMGB1 signal regulation [33], acacetin inhibits myocardial hypertrophy through the Sirt1/AMPK/PGC-1α pathway [28], attenuates aging heart failure through Sirt1-mediated activation of Sirt6/AMPK signaling pathway [30], improves doxorubicin-induced cardiomyopathy through the Sirt1/AMPK/Nrf2 signaling pathway [27] and alleviates endothelial cell damage through the Sirt1/Sirt3/AMPK mitochondrial axis and MsrA-Nrf2/Keap1 cascade [32]. In this study, we demonstrate the novel pharmacological effect that the protection of acacetin against LysoPC-induced VSMC injury was mediated by activating Sirt1/Nrf2 molecules followed by increasing p62 phosphorylation, which is different from those in other cell types.
Therapeutic validation in HFD-fed ApoE^−/−^ mice revealed the capacity of acacetin to rescue Sirt1 expression, concomitantly reducing vascular oxidative stress and apoptosis. This translational consistency bridges cellular mechanisms with pathophysiological outcomes, positioning acacetin as a pleiotropic vascular modulator and expanding its established pharmacopeia—spanning atrial fibrillation management through ion channel blockade (atrial-selective I_Kur_ channel inhibition) [21] and metabolic regulation (hepatic DPP4 inhibition) [41]. Although suppression of LysoPC-induced Ca^2+^ influx is important for the VSMCs’ protection [11,12], LysoPC-induced Ca^2+^ influx was not effectively prevented in VSMCs with acacetin, indicating that acacetin plays a critical role in vascular protection by maintaining redox homeostasis involved in Sirt1/Nrf2 pathway, but not by Ca^2+^ modulation. Our findings augment the therapeutic portfolio of acacetin. This VSMC-focused discovery completes acacetin’s cellular defense triad (cardiomyocyte-endothelial-VSMC), underscoring its unique capacity to target multiple atherosclerotic components through unified redox/Nrf2 signaling.
The Nrf2-Keap1-p62 axis constitutes an evolutionarily conserved cytoprotective system, where Nrf2 serves as a central regulator of cellular redox homeostasis and inflammatory responses through transcriptional control of antioxidant enzymes (NQO1, CAT, SOD) and detoxification proteins [42]. Under physiological conditions, Keap1 anchors Nrf2 in the cytoplasm for proteasomal degradation, while stress-induced p62 phosphorylation at Ser349 disrupts this complex, enabling Nrf2 nuclear translocation and subsequent activation of antioxidant response element (ARE)-driven genes [39,43]. Our findings demonstrate that acacetin (1 and 3 μM) protects against LysoPC-induced VSMC injury by enhancing p62 phosphorylation to competitively bind Keap1 and suppressing Keap1 expression, collectively promoting nuclear Nrf2 accumulation and upregulating cytoprotective enzymes (NQO1 and SOD1). Genetic silence of Nrf2 completely abolished these effects, corroborating its indispensable role—a phenomenon consistent with our previous observations in endothelial cells and cardiomyocytes [27,44].
Crucially, the Nrf2 activation is governed by upstream Sirt1, an NAD^+^-dependent deacetylase that integrates metabolic sensing with stress adaptation through targets spanning PGC-1α to NF-κB [45]. Activating Sirt1/Nrf2/p62 signaling may inhibit oxidative stress, inflammation, and autophagy through mediating autophagy-dependent ferroptosis [46]. Clinically relevant Sirt1 deficiency evidenced by lower serum levels in coronary atherosclerosis patients [47] was recapitulated in LysoPC-challenged VSMCs and HFD-fed ApoE^−/−^ aortic tissues, both rescued by acacetin treatment. Sirt1 silencing abolished acacetin-mediated Nrf2/p62 activation, mechanistically linking Sirt1 to the Nrf2 pathway, a regulatory axis we previously characterized in cardiac [28] and endothelial [31] systems.
Although these results are encouraging, there are limitations that should be noted. Firstly, the specific upstream kinases for p62 phosphorylation, the acetylation profiles of Nrf2 and p62, how exactly they are regulated by Sirt1, and the impact of acacetin on these proteins need to be further clarified. Second, this study relied mainly on cellular tools like siRNA to demonstrate pathway associations; therefore, future studies using Sirt1 conditional knockout mice are needed for in vivo verification. Finally, whether the Sirt1/Nrf2/p62 axis operates in the same manner in advanced human plaques accompanied by severe calcification and macrophage infiltration, as well as the clinical targeted delivery strategies for acacetin, remain to be studied.
5. Conclusions
These findings position acacetin as a unique modulator, concurrently addressing oxidative stress (via Sirt1/Nrf2/p62) and metabolic dysregulation (through Sirt1’s deacetylation network). With demonstrated efficacy in reducing VSMC apoptosis and ROS production in preclinical models, acacetin emerges as a promising therapeutic drug candidate for atherosclerotic plaque stabilization through multimodal vascular protection.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Goncalves I. Edsfeldt A. Ko N.Y. Grufman H. Berg K. Bjorkbacka H. Nitulescu M. Persson A. Nilsson M. Prehn C. Evidence supporting a key role of Lp-PLA 2-generated lysophosphatidylcholine in human atherosclerotic plaque inflammation Arterioscler. Thromb. Vasc. Biol.2012321505151210.1161/ATVBAHA.112.24985422499993 · doi ↗ · pubmed ↗
- 2Dohi T. Miyauchi K. Ohkawa R. Nakamura K. Kurano M. Kishimoto T. Yanagisawa N. Ogita M. Miyazaki T. Nishino A. Increased lysophosphatidic acid levels in culprit coronary arteries of patients with acute coronary syndrome Atherosclerosis 201322919219710.1016/j.atherosclerosis.2013.03.03823664202 · doi ↗ · pubmed ↗
- 3Law S.H. Chan M.L. Marathe G.K. Parveen F. Chen C.H. Ke L.Y. An Updated Review of Lysophosphatidylcholine Metabolism in Human Diseases Int. J. Mol. Sci.201920114910.3390/ijms 2005114930845751 PMC 6429061 · doi ↗ · pubmed ↗
- 4Kume H. Harigane R. Rikimaru M. Involvement of Lysophospholipids in Pulmonary Vascular Functions and Diseases Biomedicines 20241212410.3390/biomedicines 1201012438255229 PMC 10813361 · doi ↗ · pubmed ↗
- 5Corrêa R. Silva L.F.F. Ribeiro D.J.S. Almeida R.D.N. Santos I.O. Corrêa L.H. de Sant’Ana L.P. Assunção L.S. Bozza P.T. Magalhães K.G. Lysophosphatidylcholine Induces NLRP 3 Inflammasome-Mediated Foam Cell Formation and Pyroptosis in Human Monocytes and Endothelial Cells Front. Immunol.201910292710.3389/fimmu.2019.0292731998284 PMC 6962110 · doi ↗ · pubmed ↗
- 6Matsumoto T. Kobayashi T. Kamata K. Role of lysophosphatidylcholine (LPC) in atherosclerosis Curr. Med. Chem.2007143209322010.2174/09298670778279389918220755 · doi ↗ · pubmed ↗
- 7Kurano M. Kano K. Dohi T. Matsumoto H. Igarashi K. Nishikawa M. Ohkawa R. Ikeda H. Miyauchi K. Daida H. Different origins of lysophospholipid mediators between coronary and peripheral arteries in acute coronary syndrome J. Lipid Res.20175843344210.1194/jlr.P 07180328007846 PMC 5282959 · doi ↗ · pubmed ↗
- 8Kurano M. Suzuki A. Inoue A. Tokuhara Y. Kano K. Matsumoto H. Igarashi K. Ohkawa R. Nakamura K. Dohi T. Possible involvement of minor lysophospholipids in the increase in plasma lysophosphatidic acid in acute coronary syndrome Arterioscler. Thromb. Vasc. Biol.20153546347010.1161/ATVBAHA.114.30474825425621 · doi ↗ · pubmed ↗