Quantitative microbial risk assessment of antibiotic resistance genes and mobile genetic elements in orchard soils across South Korea
Raan Shin, Seunggyun Han, Jaeyoung Ro, Sujin Lee, Song-Hee Ryu, Hor-Gil Hur, Hanseob Shin

TL;DR
This study shows that orchard soils in South Korea contain antibiotic resistance genes, and farm workers are at risk of ingesting them through soil contact.
Contribution
The study introduces a framework combining resistome profiling, microbial networks, and QMRA to assess occupational risks of antibiotic resistance gene exposure in orchard soils.
Findings
297 ARGs and 52 MGEs were detected in orchard soils, with eight core genes significantly enriched in these soils.
aac(3)-VIa posed the highest ingestion risk for orchard farmers, with ~29 annual events per person.
Exposure to ARGs in orchard soils may enrich resistance in the gut microbiome of farm workers, increasing treatment complications.
Abstract
Antibiotic resistance is a global health crisis, but environmental pathways of resistance dissemination to farm workers remain poorly understood. Agricultural soils represent critical but underexplored reservoirs of antibiotic resistance genes (ARGs) and mobile genetic elements (MGEs), particularly in orchards where antibiotics such as streptomycin and oxytetracycline are widely used for fire blight control. Here, we conducted a nationwide investigation of orchard soils in South Korea, integrating high-throughput qPCR, 16S rRNA gene sequencing, and quantitative microbial risk assessment (QMRA). We detected 297 ARGs and 52 MGEs, with eight core genes [aac(3)-VIa, tetL, aadE, sul1, qacH_351, tnpA-1, IS6100, and intI1] significantly enriched in orchard soils but absent in non-orchard soils, such as national parks or mountain soils. Aminoglycoside- and tetracycline-resistance genes were…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
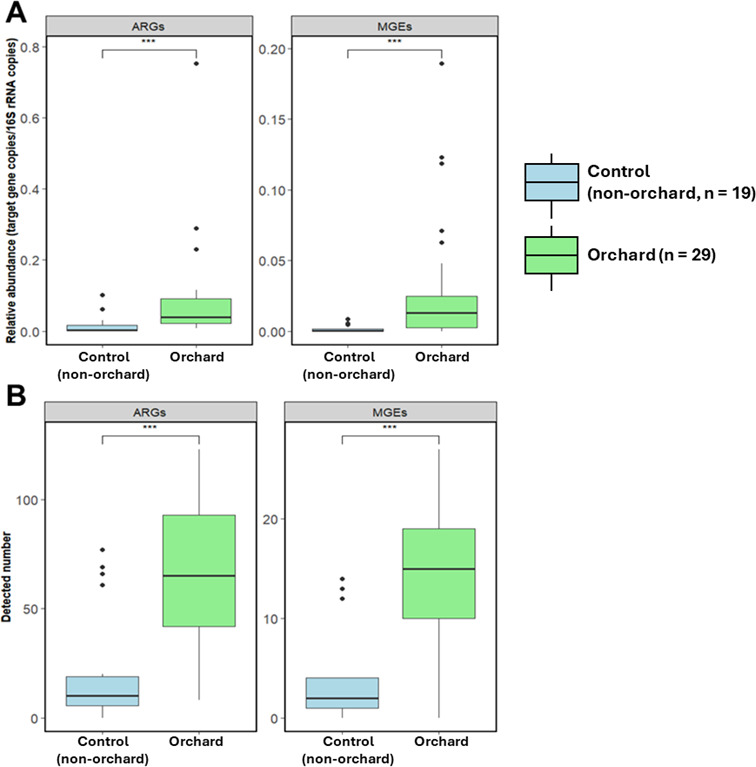 Fig 1
Fig 1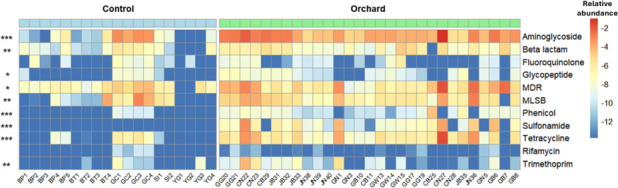 Fig 2
Fig 2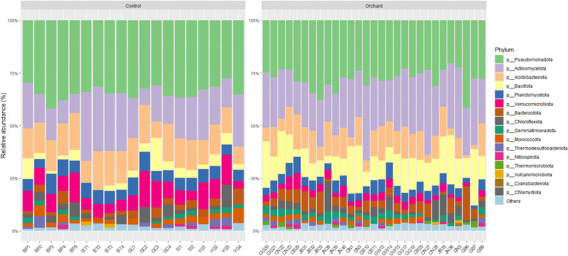 Fig 3
Fig 3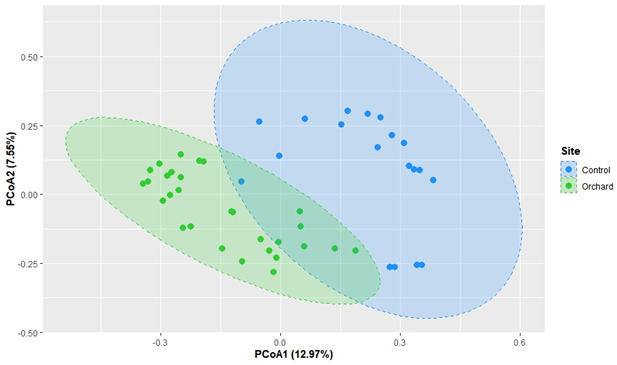 Fig 4
Fig 4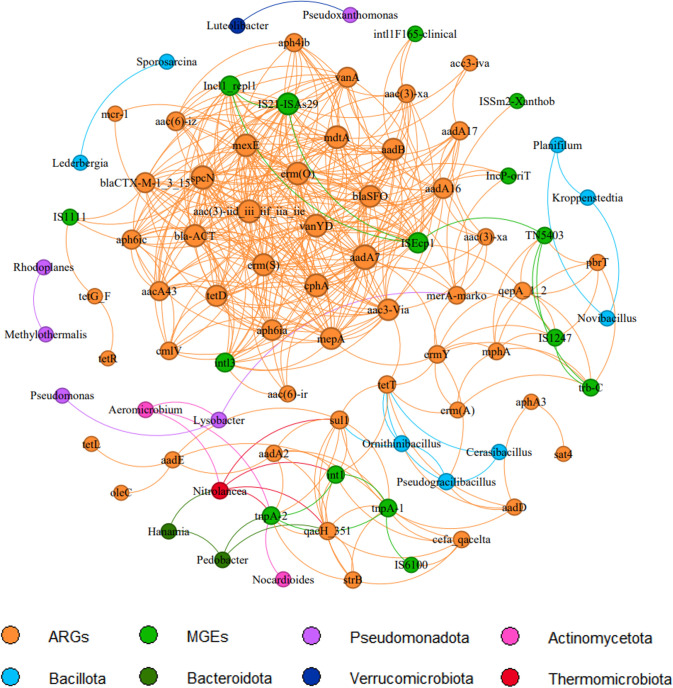 Fig 5
Fig 5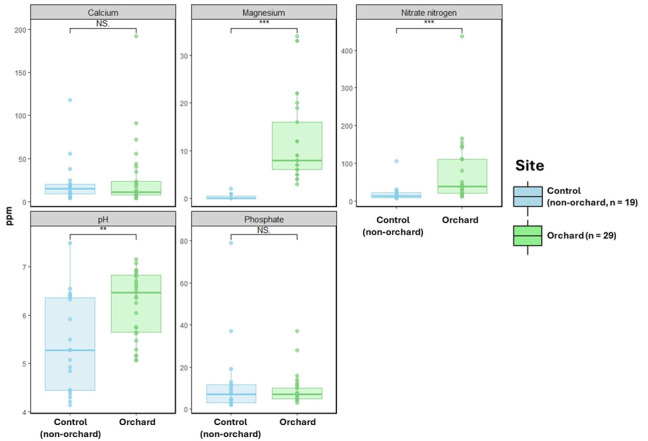 Fig 6
Fig 6| Target gene | Daily ingested gene dose, | ||
|---|---|---|---|
|
| 1.99 (1.3–3.06) | 0.52 (0.43–0.60) | 29.38 (24.51–34.2) |
|
| 0.77 (0.26–3.26) | 0.18 (0.10–0.29) | 10.41 (5.7–16.53) |
|
| 0.49 (0.17–1.73) | 0.15 (0.08–0.25) | 8.78 (4.56–14.25) |
| 0.43 (0.18–1.26) | 0.17 (0.09–0.27) | 9.5 (5.13–15.39) | |
|
| 0.17 (0.10–0.25) | 0.09 (0.06–0.13) | 5.38 (3.42–7.41) |
|
| 0.43 (0.13–1.63) | 0.13 (0.07–0.22) | 7.62 (4.0–12.54) |
|
| 0.30 (0.11–1.00) | 0.12 (0.06–0.20) | 6.57 (3.42–11.4) |
|
| 0.60 (0.18–1.81) | 0.18 (0.08–0.29) | 10.2 (4.56–16.53) |
- —National Research Foundation of Koreahttp://dx.doi.org/10.13039/501100003725
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsPharmaceutical and Antibiotic Environmental Impacts · Antibiotic Use and Resistance · Pesticide and Herbicide Environmental Studies
INTRODUCTION
The rising use of antibiotics significantly contributes to the emergence and spread of antibiotic resistance (AR), posing a growing threat to both human and animal health (1). Despite these concerns, the development and continued use of antibiotics remain indispensable for modern medicine. In addition to their clinical applications, antibiotics are widely consumed in agriculture for crop protection, animal husbandry, and livestock production (2). It has been well documented that agricultural antibiotic usage has substantially contributed to the proliferation of antibiotic resistance genes (ARGs) (3). Moreover, beyond the direct use of antibiotics, agricultural practices such as manure and biosolid application have markedly increased the prevalence of ARGs in soils and water, thereby facilitating the persistence and dissemination of antibiotic-resistant bacteria (ARB) (2, 4). For instance, elevated ARG levels have also been observed in greenhouse soils following the manure application (5).
In orchard systems, antibiotics have become essential for disease control, particularly against fire blight, a bacterial disease severely affecting apple and pear trees. In South Korea, streptomycin, oxolinic acid, and oxytetracycline are routinely applied during the blooming season to suppress bacterial infections (6), which occur more frequently in orchards compared to other agricultural crop systems (7). Such prolonged and repeated applications may accelerate the emergence of ARB (8). Furthermore, continuous agricultural inputs could promote the enrichment and mobilization of ARGs through mobile genetic elements (MGEs), increasing the potential for transmission via pathogens and subsequent exposure to humans, particularly orchard workers (9).
Quantitative microbial risk assessment (QMRA) is needed to protect the health of workers in agricultural environments. Since risk assessment estimates the likelihood of disease caused by infections, the widespread use of antibiotics calls attention to the urgent need to evaluate human exposure to antibiotic resistance in the environment (10). Effective risk assessment requires the identification and continuous surveillance of ARGs (11, 12). Then, QMRA provides a structured framework to quantify potential exposure to antibiotic resistance in specific environments and to evaluate the associated health risks (13). A study in the United States assessed the risk of ARG and MGE exposure through contaminated wells (13), while another study in India used QMRA to evaluate the risk of exposure to β-lactam resistance genes in water and street food (14). Despite increasing concerns, systematic risk assessment has not been conducted in agricultural environments in South Korea.
Thus, in South Korea, where antibiotics are widely applied in orchards, understanding the dynamics of ARGs is essential for QMRA to protect agricultural workers. We conducted a nationwide assessment of ARGs and MGEs in orchard soils and further applied QMRA to evaluate potential exposure risks to workers. This study could provide critical risk assessment information on the potential human health impacts associated with ARG exposure in agricultural environments.
RESULTS AND DISCUSSION
Resistome and mobilome analysis in orchard samples
A total of 297 ARGs and 52 MGEs were identified in orchard soil samples. The relative abundances of ARGs and MGEs were significantly higher in orchard soils compared to controls (P < 0.001, Fig. 1A). Similarly, the number of detected ARGs and MGEs was also significantly greater in orchard samples than in controls (P < 0.001, Fig. 1B). In particular, with the exception of fluoroquinolone- and rifamycin-resistance genes, all other ARG classes showed significantly higher relative abundances in orchard samples compared to controls (P < 0.05, Fig. 2). Notably, aminoglycoside-resistance genes exhibited the highest enrichment across orchard soils, showing their dominant role in these environments (P < 0.001). Other ARG classes, including phenicol, sulfonamide, and tetracycline, were also significantly more abundant in orchard samples (P < 0.001). β-Lactam, trimethoprim, and macrolide-lincosamide-streptogramin (MLSB) resistance genes were moderately enriched (P < 0.01), while multidrug resistance and glycopeptide resistance genes were detected at significantly higher levels in orchard soils than in controls (P < 0.05).
*Comparison of the abundance and diversity of antibiotic resistance genes (ARGs) and mobile genetic elements (MGEs) between orchard (n = 29) and control (non-orchard, n = 19) samples. (A) Relative abundance of ARGs and MGEs in orchard and control samples. (B) Detected numbers of ARGs and MGEs in orchard and control samples. Statistical significance was determined using the Wilcoxon test. **P < 0.001.
*A heatmap for the distribution of ARG types in orchard and control samples. The legend values represent log10 (relative abundance, target gene copies/16S rRNA gene copies). In the legend, red and blue indicate higher and lower relative abundance, respectively. Green and blue bars above the heatmap indicate orchard and control samples, respectively. Statistical significance was determined using the Wilcoxon test. *P < 0.05, **P < 0.01, **P < 0.001. MDR, multidrug resistance.
These observations align with previous studies in agricultural environments, such as greenhouse soils, which also exhibited higher ARG diversity and abundance compared to non-agricultural soils (15). Furthermore, long-term agricultural activities, including manure application and antibiotic treatments, contributed to the enrichment and dissemination of ARGs by exerting selective pressure on soil microbiomes (5). In orchard systems, streptomycin (an aminoglycoside) and oxytetracycline (a tetracycline) are among the most frequently used antibiotics for the management of fire blight (9). The dominant enrichment of aminoglycoside-resistance genes and the significantly elevated levels of tetracycline-resistance genes observed in our orchard soils are therefore consistent with the types of antibiotics commonly applied in these environments. This suggests that the continued use of streptomycin and oxytetracycline in orchards may be a major driver of ARG persistence and proliferation, reflecting direct selective pressure on the soil resistome.
Identification of core ARGs and MGEs in orchard soils
Linear discriminant analysis effect size (LEfSe) analysis identified eight core genes [five ARGs: aac(3)-VIa, tetL, aadE, sul1, and qacH_351, and three MGEs: tnpA-1, IS6100, and intI1] (Fig. S1A). Notably, a transposase gene, tnpA-1, was detected in 26 orchard samples, representing the most frequently identified core MGE, while aac(3)-VIa was detected in 28 orchard samples. Except for these two genes, the remaining six core genes (tetL, aadE, sul1, qacH_351, IS6100, and intI1) were absent in all control (non-orchard) soil samples. The concentration of tnpA-1 1 (0–0.099 copies per 16S rRNA gene copies) was the highest among MGEs and was significantly enriched in manure-amended soils compared to control soils (Fig. S1B).
These findings are consistent with previous evidence that tnpA proliferates in manure-treated agricultural environments (16). Likewise, the relative abundances of tnpA and IS6100 increased under high antibiotic concentrations, further supporting their responsiveness to anthropogenic selective pressure (17). Another MGE, intI1, also serves as a well-established indicator of ARG dissemination due to its key role in driving the horizontal transfer of multiple resistance determinants (11). Among ARGs, aac(3)-VIa exhibited the highest relative abundance (0–0.058 copies per 16S rRNA gene copy) across 28 orchard soil samples (Fig. S1B), suggesting its value as an indicator gene for monitoring ARG contamination in agricultural soils. Importantly, aac(3)-VIa is commonly carried on IncA/C plasmids and frequently associated with intI1, indicating a strong potential for horizontal gene transfer (HGT) (18). It has been reported in Escherichia coli from cattle feces and poultry, suggesting its mobility and dissemination across agricultural and environmental settings (18).
The aminoglycoside-resistance gene, aadE, has been detected in agricultural soils (19). Its presence in orchard soils is consistent with streptomycin use for fire blight management, indicating the selective pressure that drives the proliferation of aminoglycoside resistance in these environments. Similarly, the abundance of the tetracycline-resistance gene, tetL, has been shown to increase under oxytetracycline exposure and is frequently detected in Enterococcus faecalis from tetracycline-contaminated pig manure (20). The enrichment of tetL in orchard soils thus reflects the intensive use of oxytetracycline in fruit production. Together, the detection of aadE and tetL establishes a direct link between agricultural antibiotic usage (streptomycin and oxytetracycline) and the dominance of aminoglycoside- and tetracycline-resistance genes in orchard soils.
The sulfonamide resistance gene, sul1, has been frequently reported in agricultural environments (21), while qacH_351, a quaternary ammonium compound resistance gene, has been enriched in hospital wastewater and urban rivers (22). Moreover, qacH_351 abundance increased under polymyxin B treatment, suggesting that antibiotic pressure may further contribute to its environmental proliferation.
These findings demonstrate that the identified core genes in orchard soils serve as potential indicator genes for monitoring AR contamination. The enriched aminoglycoside- and tetracycline-resistance genes, in particular, herald the direct impact of streptomycin and oxytetracycline application in orchards, emphasizing the role of antibiotic pressure in shaping the soil resistome.
Bacterial community structure in orchard soils
Bacterial taxa in orchard and control soils were identified at the phylum level. The composition of major phyla (relative abundance >1%) is compared in Fig. 3. The distribution of the five dominant phyla differed markedly between orchard and control samples. In orchard soils, Pseudomonadota was the most abundant phylum, accounting for 19.9%–40.9% of the community, followed by Actinomycetota (13.4–38.2%), Bacillota (2.9%–25.3%), Acidobacteriota (1.3%–14.6%), and Bacteroidota (1.1%–15.7%) (Table S1). In contrast, the control soils were dominated by Pseudomonadota (23.3%–37.3%), Actinomycetota (6.54%–30.1%), Acidobacteriota (7.26%–17.6%), Verrucomicrobiota (3.1%–14.3%), and Planctomycetota (3.8%–8.5%) (Table S1). Statistical comparison revealed that Bacillota, Bacteroidota, and Actinomycetota were significantly more abundant in orchard soils than in controls (Wilcoxon matched-pair test, P < 0.05), whereas Pseudomonadota, Acidobacteriota, Verrucomicrobiota, and Planctomycetota were significantly enriched in control soils (P < 0.05) (Fig. S2).
Stacked bar plots for the relative abundance of bacterial phyla (>1% of total community) across orchard and control soil samples. The left and right panels represent control and orchard soils, respectively. Phyla with <1% relative abundance are grouped as others.
Among the dominant phyla, Actinomycetota and Bacillota are strongly associated with the presence and mobility of ARGs (23). Previous studies have identified Bacillota as a dominant phylum under long-term antibiotic selection pressure (24). Members of Bacillota, particularly the genera Streptococcus, Enterococcus, and Staphylococcus, are known to be major contributors to the horizontal gene transfer of ARGs through plasmids and MGEs under antibiotic pressure (25). Similarly, Actinomycetota, characterized by their high G + C content (26), are well recognized for producing diverse bioactive metabolites, including antibiotics, anticancer compounds, and enzymes (27). Actinomycetes isolated from various soil environments often exhibit resistance to an average of seven to eight antibiotics, reflecting their intrinsic resistance mechanisms and adaptive strategies in competitive soil ecosystems (28).
β-Diversity analysis further revealed that microbial communities from orchard soils clustered closely together while being clearly separated from control soils (R^2^ = 0.14, P < 0.001) (Fig. 4). LEfSe analysis identified 34 genera with LDA scores greater than 3 that were significantly enriched in orchard samples compared to controls (Fig. S3), indicating distinct microbial assemblages associated with agricultural environments. A detailed discussion of the bacterial community analysis is provided in the supplemental material.
Principal coordinate analysis (PCoA) of bacterial community composition in orchard and control soils. PCoA based on Bray–Curtis distances was performed using permutational multivariate analysis of variance with adonis function in RStudio. Each point represents the bacterial community of an individual soil sample, with green and blue indicating orchard and control soil samples, respectively. Ellipses represent the 95% confidence intervals for each group. Orchard and control soils formed distinct clusters (R² = 0.13985, P < 0.001), indicating significant differences in community composition.
Relationships among bacterial community, ARGs, and MGEs
Eight bacterial hosts, potentially carrying ARGs and MGEs, were identified through network analysis based on Spearman correlations (Fig. 5). The network analysis generated 84 nodes and 430 edges, representing significant associations between bacterial genera, MGEs, and ARGs. The eight bacterial genera showed strong correlations with multiple ARGs (ermY, tetT, aadD, sul1, merA, and cqacH_351) and MGEs (int1 and tnpA-2). Among these, Nitrolancea sp. exhibited the highest number of associations, being significantly correlated with four resistance-associated elements (qacH_351, sul1, int1, and tnpA-2), suggesting its potential importance as a key mediator in ARG dissemination. Pseudogracilibacillus sp. showed positive correlations with ermY, tetT, and aadD, while Ornithinibacillus sp. was associated with tetT, aadD, and a core gene, sul1. Pedobacter sp. was significantly correlated with aadD and another core gene, qacH_351. In addition, three genera, Aeromicrobium sp., Cerasibacillus sp., and Nocardioides sp., were significantly correlated with the mobile element tnpA-2, suggesting their potential involvement in transposon-mediated ARG transfer. Notably, Pseudogracilibacillus sp., which was not detected in the control samples, showed correlations with multiple ARGs.
Network analysis linking antibiotic resistance genes (ARGs), mobile genetic elements (MGEs), and bacterial genus. Co-occurrence networks were constructed using Spearman’s correlation (R > 0.6, P < 0.05). Nodes represent ARGs (orange), MGEs (green), or bacterial genera grouped by phylum (each color is indicated in the legend). Edges denote significant positive correlations between nodes.
A previous study reported that Pseudogracilibacillus sp. is capable of surviving under antibiotic-treated conditions and was able to persist even after the thermophilic composting phase (29). Furthermore, this genus has been identified as a dominant taxon in chicken manure compost (30), suggesting its strong ecological resilience in agricultural environments. Similarly, Pedobacter spp. are resistant to multiple antibiotics and have been predominantly found in an aminoglycoside-resistant bacterial isolate collection (31).
Influence of soil physicochemical characteristics on bacterial communities and ARGs
Orchard soils exhibited distinct physicochemical characteristics compared to control soils, with significantly higher concentrations of nitrate (NO^3−^) and magnesium (Mg^2+^) (P < 0.001) and elevated pH (P < 0.01), while calcium (Ca^2+^) and phosphate (PO_4_^3−^) showed no significant differences (Fig. 6). Spearman correlation analysis further revealed that several enriched bacterial genera were significantly associated with soil parameters, with pH showing the strongest negative correlations. Notably, genera such as Nitrolancea, Nocardioides, and Aeromicrobium were positively correlated with NO_3_^−^ and Mg^2+^ and also linked to ARGs and MGEs (Fig. S4). Detailed analyses and discussion of these correlations are provided in the supplemental material.
*Comparison of the concentrations of four soil ions (nitrate, magnesium, calcium, and phosphate) and pH between orchard and control samples. Statistical significance was determined using the Wilcoxon test. **P < 0.01, **P < 0.001.
Risk assessment of core genes in orchard through QMRA
QMRA was conducted to assess the potential risks associated with the ingestion of ARGs and MGEs by orchard farmers. The analysis focused on unintentional ingestion of soil particles carrying resistance genes, with the objective of quantifying long-term exposure risks linked to core ARGs and MGEs. Considering gene ingestion as a direct exposure pathway, this study points out the importance of soil contact as a critical route for ARG transmission in agricultural environments (orchard in this study).
Among ARGs, aac(3)-VIa showed the highest daily ingested gene dose (1.99 copy numbers/day, 95% confidence interval [CI]: 1.3–3.06) and the highest daily probability of gene ingestion (0.52, 95% CI: 0.43–0.60) (Table 1). In terms of annual exposure, aac(3)-VIa also presented the highest risk, with an estimated 29.38 ingestion events per farmer per year (95% CI: 24.51–34.20) (Table 1). This was followed by qacH_351 (10.20 cases/year), IS6100 (9.50 cases/year), tetL (8.78 cases/year), intI1 (7.62 cases/year), sul1 (6.57 cases/year), and aadE (5.38 cases/year). For MGEs, tnpA-1 exhibited the highest estimated risk at 10.41 cases/year (95% CI: 5.70–16.53), followed by IS6100 and intI1. Such MGE-mediated genes represent significant pathways of potential exposure in orchard soils.
Network-based correlation analysis (see Fig. 5) revealed strong associations between ARGs, MGEs, and specific bacterial genera, indicating that these taxa may act as carriers and facilitators of resistance dissemination. Given the frequent and unintentional soil contact by orchard farmers, identifying bacterial genera that harbor core ARGs is crucial for understanding potential transmission pathways of resistance. Although ingestion of ARGs and MGEs may not directly result in immediate illness, the possibility of HGT within the human gut microbiome poses an indirect significant public health concern (13). This study introduces a framework for evaluating ARG-related risks in orchard environments by integrating quantitative estimates of gene ingestion with microbial interaction networks, thereby improving understanding of how resistance may persist and spread in agricultural ecosystems.
Conclusions
In this study, we applied QMRA to agricultural environments as a means to evaluate human health risks from ARGs and MGEs. While the clinical aspects of antibiotic resistance are well recognized, environmental exposures, particularly through direct soil contact in orchards, remain unexplored. Indeed, gene ingestion via soil represents an exposure pathway for orchard farmers, driving the need to integrate QMRA into environmental monitoring and management frameworks.
To fill this gap, we conducted a nationwide investigation of orchard soils in South Korea, identifying 297 ARGs and 52 MGEs, including eight core genes strongly linked to streptomycin and oxytetracycline use. Distinct microbial communities enriched in orchards were correlated with ARGs, MGEs, and soil physicochemical factors. QMRA revealed that aminoglycoside-resistance gene, aac(3)-VIa, posed the highest annual exposure risk (~29 ingestion events per farmer), followed by qacH_351, tetL, and tnpA-1. By integrating comprehensive approaches (resistome profiling, microbial network analysis, and QMRA), this study could establish a framework for quantifying ARG-related risks in agroecosystems and contribute to developing strategies to mitigate resistance dissemination and protect farmer health.
MATERIALS AND METHODS
Description of sampling sites and sample collection
A total of 29 orchard soil samples were collected in 2023 from eight provinces across South Korea. To provide a representative nationwide assessment, orchard sites were selected across South Korea, covering a broad range of regions. Orchard sites were selected based on the orchard database provided by the Rural Development Administration. Sampling was conducted in March, prior to the blooming season. During site visits, some orchards had already received manure or antibiotic applications, while others were in the process of applying them. For orchard sites that were no longer accessible at the time of sampling, nearby orchards were sampled after confirming with farmers that agricultural activities were being conducted. However, detailed information regarding the quantity and duration of antibiotic or fertilizer application was not available.
In addition, 19 non-orchard soil samples were collected as controls from mountainous areas and provincial parks that were not influenced by agricultural activities. Detailed information on all sampling sites is provided in Table S2. Soil samples from each site were collected as composite samples from a depth of 5–15 cm. All samples were transported to the laboratory under cold conditions and stored at −20°C until further analysis.
Analysis of soil physicochemical parameters
The dried soils were sieved through a 2 mm mesh prior to downstream analyses. The concentrations of Mg^2+^, Ca^2+^, NO^3−^, PO_4_^3−^, and pH were measured using a Rapid-d PIA-001 ion analyzer (Technell, South Korea) following the manufacturer’s instructions. For pH determination, 5 g of soil was suspended in 25 mL of sterile distilled water, shaken for 1 h, and measured using a Star A211 Benchtop pH Meter (Thermo Fisher).
DNA extraction
DNA was extracted from 0.5 g of soil using the DNeasy PowerSoil kit (Qiagen, Hilde, Germany) according to the manufacturer’s instructions. The concentration of the extracted DNA was measured using a Nanodrop spectrophotometer (MicroDigital Co., Korea). Extracted DNA samples were stored at −20°C for downstream analysis.
High-throughput quantitative PCR of antibiotic resistance gene and mobile genetic elements
High-throughput quantitative PCR (HT-qPCR) was conducted using the SmartChip qPCR program (v.2.7.0.1; Wafergen Biosystem, USA) to quantify target genes in the samples. A total of 382 primers were designed and validated to amplify 319 ARGs, 57 MGEs, and 16S rRNA genes (32). The performance of the HT-qPCR array has been experimentally validated, showing amplification efficiency between 90% and 110%, detection limits as low as 1–10 gene copies/reaction, a linear dynamic range spanning approximately six to seven orders of magnitude, and standard curves with R^2^ > 0.99 (32). The 319 ARG primer sets include major antibiotic classes such as aminoglycosides, β-lactams, fluoroquinolones, glycopeptides, MLSB, phenicols, rifampicins, sulfonamides, multidrug ARGs, tetracyclines, and trimethoprim. For the 57 MGEs, insertional, integrase, plasmid, and transposase were included.
Each PCR mixture contains 1× LightCycler 480 SYBR Green I Master Mix (Roche Inc., Basel, Switzerland), 5 ng/µL of DNA template, 500 nM of reverse and forward primers (32), and nuclease-free PCR-grade water. Amplification was performed using the following protocol: initial denaturation at 95°C for 10 minutes, followed by 40 cycles of denaturation at 95°C for 30 seconds, and annealing at 60°C for 30 seconds. Melting curve analysis was provided using the SmartChip qPCR program (v.2.7.0.1, Wafergen Biosystem). All qPCR reactions were performed in triplicate, and only the positive runs in triplicates were used for analysis. Reactions with multiple melting peaks or amplification efficiencies outside the range of 1.8–2.2 were discarded. A threshold cycle of 31 was considered the detection limit. Gene copy numbers were calculated using the following equation (33): gene copy number = . The copy numbers of ARGs and MGEs were normalized by those of 16S rRNA to determine the relative abundance.
Risk assessment
The QMRA approach was used to assess the risk associated with the potential ingestion of ARGs and MGEs by orchard farm workers. QMRA was conducted in the following sequence (34): (i) hazard identification, (ii) exposure assessment, (iii) dose–response assessment, and (iv) risk characterization.
Hazard identification
LEfSe algorithm was used to identify core genes (35) and to compare orchard soils with control (non-orchard) soils in South Korea. In this study, core genes were defined as ARGs and MGEs that were consistently and significantly enriched across multiple orchard soil samples relative to control soils. Core genes significantly enriched in orchard soils were considered indicators of resistance contamination in agricultural environments. Since ARGs and MGEs have the potential to pose health risks (13, 36, 37), they were defined as potential hazards in orchard soils in this study.
Exposure assessment
Exposure assessment was conducted to estimate the potential ingestion of ARGs and MGEs by farmers through soil contact. The model focused on unintentional ingestion of soil particles during agricultural activities. Based on similar agricultural environments (38), our study assumed that farmers ingest between 10 and 200 mg of soil per day in orchards. This range was determined from WHO and EPA guidelines, particularly EPA’s high-contact scenarios and soil-plus-dust ingestion rates for adults. The selected range (10–200 mg/day) reflects higher levels of soil exposure in labor-intensive agricultural settings, where farmers frequently come into contact with soil. The daily exposure dose (D, gene copy numbers/day) was calculated based on the gene copy numbers estimated from the relative abundance of target genes. It was obtained by multiplying the gene concentration in the soil (C, gene copy numbers/mg of soil) by the daily soil ingestion rate (R, mg/day), as shown in equation (1):
Dose–response assessment
The daily probability of gene ingestion (P, unitless) was estimated using an exponential dose–response model with a conservative coefficient r = 1^13^. Because specific dose–response parameters for ARGs and MGEs have not been experimentally validated, the assumption of r = 1 follows standard QMRA practice for highly uncertain agents. P was calculated following equation (2):
In this equation, D represents the daily dose of the gene (gene copy numbers/day) estimated in the exposure assessment, and P is the probability of the daily dose of gene ingested by farmers.
Risk characterization
Risk was characterized as the annual number of gene ingestion events for individual orchard farmers, as shown in equation (3). The parameters used in risk assessment were the previously calculated probability of the daily dose (P) and the estimated annual working days of orchard farmers (d/y) averaging about 50 days per year (range: 20–65 days) based on reported working times for major fruit crops (39). The estimated annual exposure cases (E, gene ingestion events/year) were calculated based on equation (3):
where P is the daily probability of gene ingestion, and the annual working days (d/y) represent the number of working days per year. Based on reported working times for major fruit crops (apple, pear, and grape) (39), orchard farmers worked on average 50 days per year (range: 20–65 days), consistent with typical orchard maintenance and harvest periods (40).
Analysis of bacterial community using 16S rRNA gene sequencing
For bacterial community analysis, the hypervariable region (V3–V4) of the bacterial 16S rRNA gene was amplified with primers (341F: CCTACGGGNGGCWGCAG and 805R: GACTACHVGGGTATCTAATCC). Amplicon sequencing was performed using the Illumina Miseq platform. Amplicon sequence variants (ASVs) were generated following the DADA2 (v.1.18.0) pipeline (41), which included steps for error correction, merging, and denoising. Taxonomy classification was performed by classifying ASVs using the NCBI 16S reference database with a Bayesian classifier implemented in DADA2 (v.1.18.0) (42), and downstream analyses were conducted using QIIME (v.1.9.0) (43).
Statistical analysis
Data analysis and calculations were performed by using R Studio (v.4.3.3) and Microsoft Excel 2020. The Wilcoxon test was applied to compare the relative abundances of ARGs, MGEs, ARG type, soil physicochemical parameters, and bacterial phyla between orchard and control samples, using the “ggsignif” package in R studio. Principal coordinate analysis based on Bray–Curtis distances was performed using the “vegan” package to determine the distribution of bacterial communities. Statistical significance of compositional differences was further evaluated with permutational multivariate analysis of variance using the adonis function in the vegan package. To identify core genes and bacterial genera enriched in orchard samples, the LEfSe algorithm was performed (44). The core bacterial genera identified through LEfSe were further used for network analysis and correlation with soil physicochemical parameters. Relationships between ARGs, MGEs, and the bacterial genera in orchard samples were analyzed through Spearman’s correlation. Based on Spearman coefficients (R^2^ > 0.63, P < 0.05), a co-occurrence network was constructed using Gephi.
QMRA was performed using two-dimensional Monte Carlo (2DMC) simulations, utilizing the “mc2d” package and the “fitdistrplus” package (45). The 2DMC approach separates uncertainty and variability. This study conducted 1,000 iterations in each dimension (total of 4,000,000 [2,000 uncertainty dimension × 2,000 variability dimension] Monte Carlo simulations). The gene concentration data (C) was modeled by fitting parametric distributions. C was assumed to be based on distributions defined by bootstrap procedure, reflecting uncertainty and variability. Daily ingestion rates of soil (R) were assumed to follow a uniform distribution (10–200 mg/day) to represent variability. The daily exposure dose (D) was simulated using a Poisson distribution based on the estimated exposure dose. Monte Carlo simulation outputs were summarized by calculating the arithmetic mean in the variability dimension, and then calculating the median and 95% confidence intervals across the uncertainty dimension.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Tiseo K, Huber L, Gilbert M, Robinson TP, Van Boeckel TP. 2020. Global trends in antimicrobial use in food animals from 2017 to 2030. Antibiotics (Basel) 9:1–14. doi:10.3390/antibiotics 9120918 · doi ↗
- 2Mann A, Nehra K, Rana JS, Dahiya T. 2021. Antibiotic resistance in agriculture: perspectives on upcoming strategies to overcome upsurge in resistance. Curr Res Microb Sci 2:100030. doi:10.1016/j.crmicr.2021.10003034841321 PMC 8610298 · doi ↗ · pubmed ↗
- 3Wang F, Fu YH, Sheng HJ, Topp E, Jiang X, Zhu YG, Tiedje JM. 2021. Antibiotic resistance in the soil ecosystem: a One Health perspective. Curr Opin Environ Sci Health 20:100230. doi:10.1016/j.coesh.2021.100230 · doi ↗
- 4Li Y, Kong F, Li S, Wang J, Hu J, Chen S, Chen Q, Li Y, Ha X, Sun W. 2023. Insights into the driving factors of vertical distribution of antibiotic resistance genes in long-term fertilized soils. J Hazard Mater 456:131706. doi:10.1016/j.jhazmat.2023.13170637247491 · doi ↗ · pubmed ↗
- 5Fang H, Wang H, Cai L, Yu Y. 2015. Prevalence of antibiotic resistance genes and bacterial pathogens in long-term manured greenhouse soils as revealed by metagenomic survey. Environ Sci Technol 49:1095–1104. doi:10.1021/es 504157 v 25514174 · doi ↗ · pubmed ↗
- 6Ham H, Oh GR, Park DS, Lee YH. 2022. Survey of oxolinic acid-resistant Erwinia amylovora in Korean apple and pear orchards, and the fitness impact of constructed mutants. Plant Pathol J 38:482–489. doi:10.5423/PPJ.OA.04.2022.005936221920 PMC 9561153 · doi ↗ · pubmed ↗
- 7Ha H-Y, Park S-E, You A-S, Gil G-H, Park J-E, Lee I-Y, Park K-W, Ihm Y-B. 2016. Survey of pesticide use in leaf and fruit vegetables, fruits, and rice cultivation areas in Korea. Weed Turfgrass Sci 5:203–212. doi:10.5660/WTS.2016.5.4.203 · doi ↗
- 8Mc Ghee GC, Sundin GW. 2011. Evaluation of kasugamycin for fire blight management, effect on nontarget bacteria, and assessment of kasugamycin resistance potential in Erwinia amylovora. Phytopathology 101:192–204. doi:10.1094/PHYTO-04-10-012820923369 · doi ↗ · pubmed ↗