Molecular evolution and diversity of isomerase–reductase clusters involved in the bacterial metabolism of glycosaminoglycans
Yu Nishimura, Kenji Okumura, Sayoko Oiki, Kohei Ogura, Wataru Hashimoto

TL;DR
This study explores how bacteria evolve to metabolize glycosaminoglycans, revealing diverse enzyme clusters and their roles in human colonization.
Contribution
The study identifies hybrid and distinct enzyme clusters involved in glycosaminoglycan metabolism across diverse bacterial species.
Findings
Ten cluster structures are involved in DHU metabolism among 16 possible types.
Bacteroidota have hybrid kduI–dhuD clusters, while Bacillota have dhuD–dhuI clusters.
UGL gene is frequently found in pathogenic strains with specific DHU metabolism clusters.
Abstract
Glycosaminoglycans (GAGs), comprising uronic acids and amino sugars, are widely distributed in human tissues such as the intestine and oral cavity. Various bacteria colonize these tissues by assimilating GAGs. During GAG degradation, 4-deoxy-l-threo-5-hexosulose uronate (DHU) is produced. Pectin, an abundant plant component, is also degraded into DHU. DHU is metabolized in a stepwise manner by the isomerase KduI or its nonhomologous isofunctional enzyme DhuI, followed by the reductase KduD or DhuD, belonging to the same reductase–dehydrogenase family. Previous studies have found that the genes encoding isomerase and reductase (kduI–kduD and dhuD–dhuI, respectively) are usually organized in clusters. Therefore, it was believed that the kduI–kduD and dhuD–dhuI clusters evolved independently. However, the discovery of a hybrid kduI–dhuD cluster raised questions regarding the evolution of…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
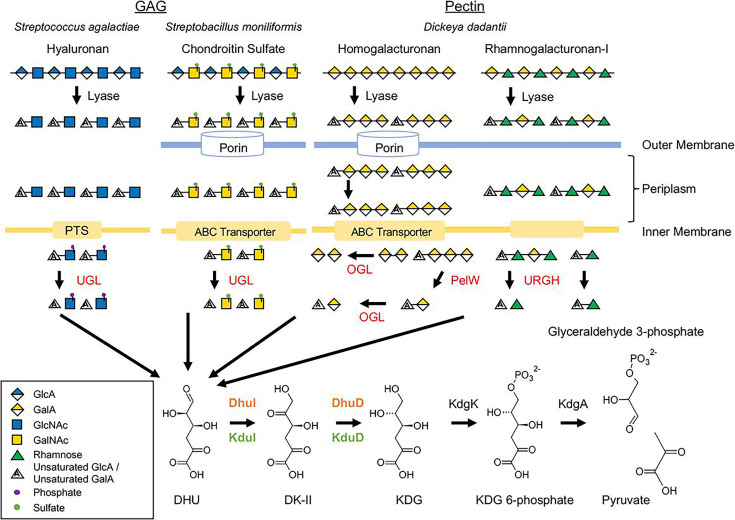 Fig 1
Fig 1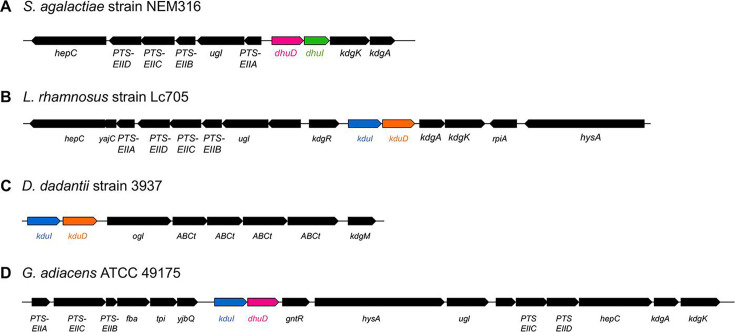 Fig 2
Fig 2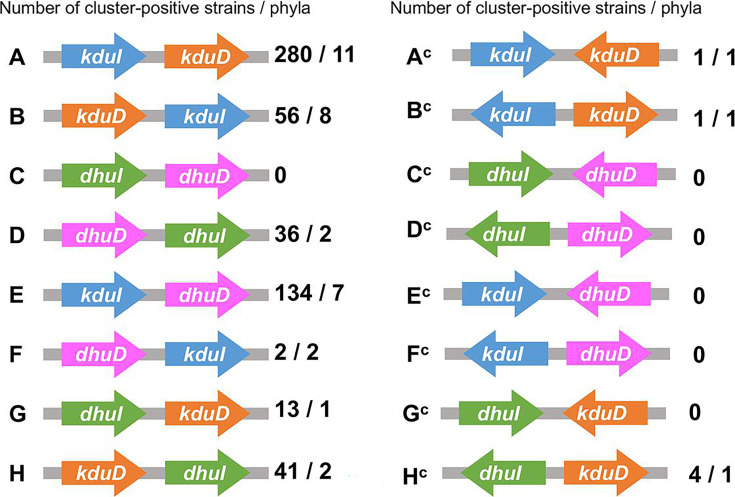 Fig 3
Fig 3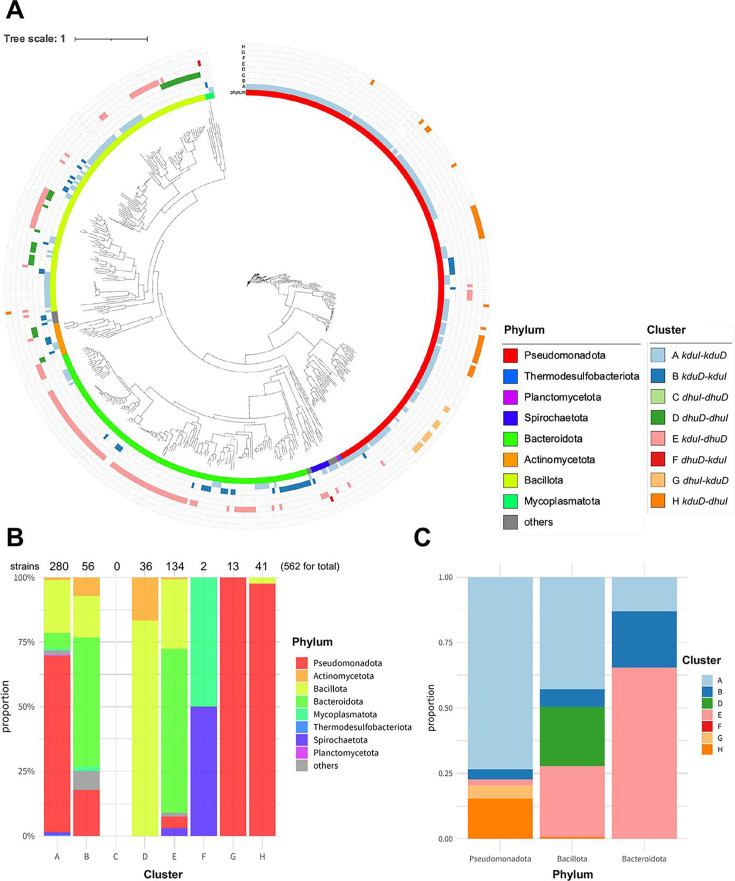 Fig 4
Fig 4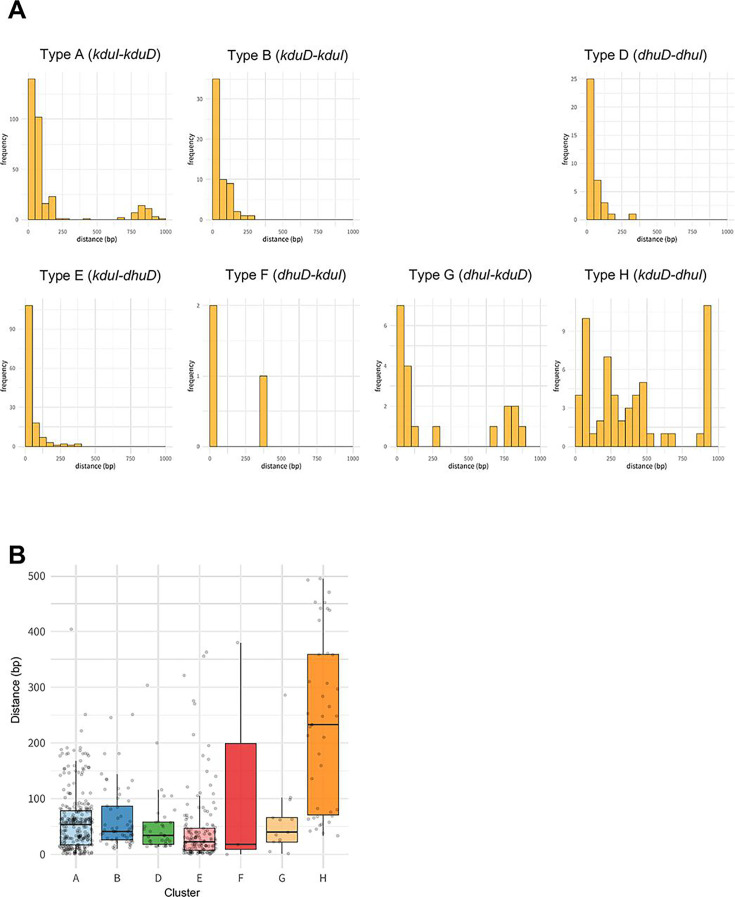 Fig 5
Fig 5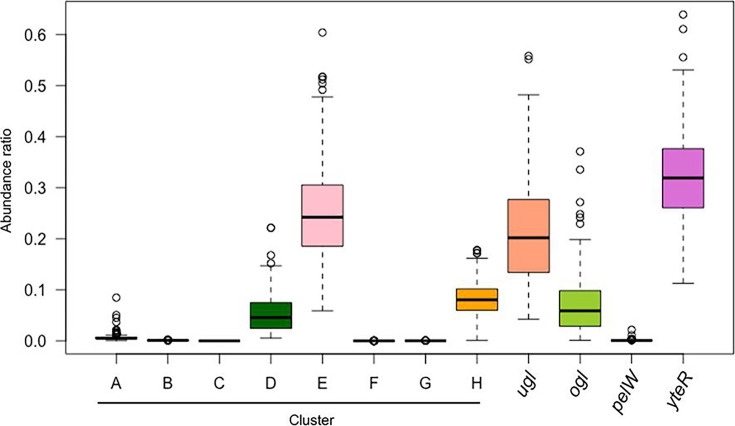 Fig 6
Fig 6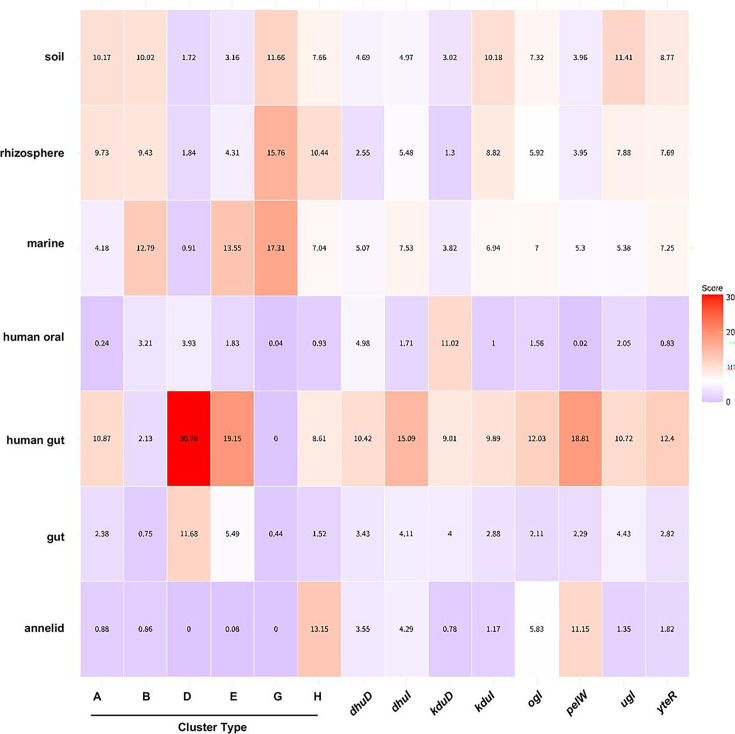 Fig 7
Fig 7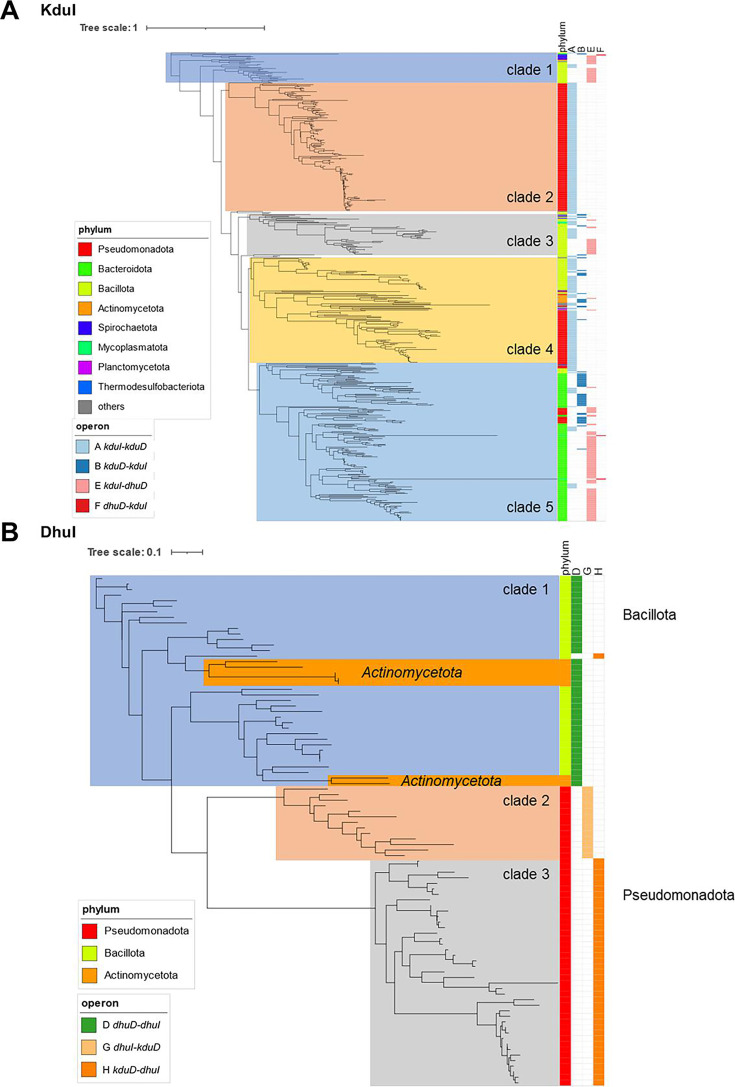 Fig 8
Fig 8- —Japan Society for the Promotion of Sciencehttp://dx.doi.org/10.13039/501100001691
- —Japan Society for the Promotion of Sciencehttp://dx.doi.org/10.13039/501100001691
- —Kyoto University Foundationhttp://dx.doi.org/10.13039/100016926
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsProteoglycans and glycosaminoglycans research · Seaweed-derived Bioactive Compounds · Enzyme Production and Characterization
INTRODUCTION
Glycosaminoglycans (GAGs) are linear polysaccharides composed of uronic acids and amino sugars that play vital roles in the extracellular matrices of animals (1). Major types of GAGs include nonsulfated hyaluronic acid (composed of glucuronic acid and N-acetylglucosamine), chondroitin sulfate and dermatan sulfate (glucuronic acid or iduronic acid and N-acetylgalactosamine with sulfation at various positions), heparin/heparan sulfate (repeating units of uronic acid and glucosamine with varying degrees of sulfation), and keratan sulfate (unique among GAGs because it contains galactose instead of uronic acid, along with N-acetylglucosamine) (2).
Specific pathogenic bacteria, such as Streptococcus pneumoniae and Streptococcus dysgalactiae, have evolved specialized gene clusters for degrading and utilizing GAGs, particularly hyaluronic acid (3–5). This process is crucial for bacterial pathogenicity because it involves the extracellular degradation of hyaluronic acid by hyaluronate lyase, followed by intracellular transport and degradation of unsaturated disaccharides. GAGs are also important for mediating interactions between the host and gut bacteria (6, 7). The unsaturated disaccharides produced by the lyase reaction are transported into the cytoplasm via the phosphotransferase system or ATP-binding cassette transporters (4, 8, 9) (Fig. 1). In Streptococcus species, disaccharides transported by the phosphotransferase system are degraded by unsaturated glucuronyl hydrolases (UGLs) belonging to glycoside hydrolase family 88 (8, 10). These UGLs degrade unsaturated disaccharides into unsaturated glucuronic acid and amino sugars. The unsaturated glucuronic acid undergoes nonenzymatic ring opening, producing 4-deoxy-l-threo-5-hexosulose uronate (DHU). In some bacteria, the enzyme KdgF facilitates this ring opening reaction enzymatically (11).
Metabolism of GAGs and pectins. Metabolism of GAGs and pectins. The names of saccharides were described according to a previous report (12).
Pectin, a heteropolysaccharide abundant in the plant cell walls, is primarily composed of α-1,4-d-galacturonic acid units and contains various neutral sugars such as rhamnose, arabinose, galactose, and xylose (13). Pectin lyases are produced by various bacteria such as the plant pathogen Dickeya dadantii, the human gut bacterium Bacteroides thetaiotaomicron, and the human pathogen Yersinia enterocolitica (14). In D. dadantii, pectin oligosaccharides produced by pectin lyase are transported into the periplasm via porins KdgM and/or KdgN (15–17), followed by passage into the cytoplasm through the ATP-binding cassette transporter TogMNAB (18, 19). In the cytoplasm, oligogalacturonate lyases (OGLs) catalyze the conversion of saturated and unsaturated digalacturonates into monogalacturonate and DHU (20) (Fig. 1). Thus, DHU is produced from GAGs and pectin. In D. dadantii, DHU is also generated from unsaturated rhamnogalacturonan by unsaturated galacturonyl hydrolase (YteR), a member of the GH105 family (21).
In Streptococcus agalactiae, DHU is isomerized by DhuI into 3-deoxy-d-glycero-2,5-hexodiulosonate (DK-II), which is further reduced by the NADH-dependent reductase DhuD to form 2-keto-3-deoxy-d-gluconate (22). The enzymes DhuD and DhuI are encoded by the cluster dhuD–dhuI in the S. agalactiae genome (Fig. 2A). The probiotic Lacticaseibacillus rhamnosus and the plant pathogen D. dadantii produce the isomerase KduI and the NADH-dependent reductase KduD, whose genes also exist in a cluster as kduI–kduD (23) (Fig. 2B and C). The genes dhuI and kduI encode enzymes that catalyze similar reactions but lack sequence homology, indicating the presence of nonhomologous isofunctional enzymes. KduI and DhuI belong to the KduI/IolB isomerase family (InterPro ID, IPR021120) and the RpiB/LacA/LacB sugar-phosphate isomerase family (IPR003500), respectively. Although KduD and DhuD belong to the short-chain dehydrogenase–reductase family (IPR002347), their sequence identity is low. For example, KduD from L. rhamnosus only exhibit 42.2% identity with DhuD from S. agalactiae. Therefore, the clusters dhuD–dhuI and kduI–kduD are believed to have evolved independently. However, a hybrid cluster kduI–dhuD was recently detected in Granulicatella adiacens, isolated from the oral cavity (Fig. 2D) (24). This discovery of a hybrid cluster raised questions about the evolutionary process of cluster formation.
Gene clusters involved in the metabolism of GAGs and pectins. GAG-metabolizing gene clusters of (A) S. agalactiae strain NEM316 (NC_004368.1), (B) L. rhamnosus strain Lc705 (NC_013199.1), (C) D. dadantii strain 3937 (NC_014500.1), and (D) G. adiacens ATCC 49175 (GG694015.1). The term “ABCt” refers to genes that encode components of an ABC transporter. The illustrations were created using the drawGeneArrows3 tool, developed by Dr. Yoshiyuki Ohtsubo from Tohoku University (https://www.ige.tohoku.ac.jp/joho/).
The ability of microbes to metabolize diverse saccharides provides a significant evolutionary advantage, enabling them to thrive in environments in which preferred carbon sources are scarce. This pressure has led to the development of highly specialized operons for different sugars. Kaznadzey et al. (25) reported that approximately 50% of studied genes are co-localized in bacterial genomes with other carbohydrate metabolism genes and that the preference toward the co-localization of carbohydrate metabolism genes varies between 40% and 76% for bacterial taxa. Bacteria can acquire entire operons or gene clusters involved in saccharide metabolism from other bacteria through horizontal gene transfer (26). Duplication of genes followed by divergence of the duplicated copies can lead to novel genes with altered substrate specificities or catalytic activities within a saccharide metabolic pathway, which are then co-opted into existing or novel operons or gene clusters (27).
Although nonhomologous isofunctional enzymes are well documented, only a few studies have investigated the formation of gene clusters encoding nonhomologous isofunctional enzymes catalyzing the same sequential reaction. Furthermore, there has been limited research on the diversity of cluster structures across bacterial genomes. Our investigation focused on cluster formation and structure, particularly examining clusters related to the metabolism of GAGs. Hence, this study aimed to classify cluster structures across bacterial genomes and highlight the metabolism of GAGs and the intriguing formation of clusters encoding nonhomologous isofunctional enzymes. Understanding these complex genetic arrangements provides valuable insights into the evolutionary pressures shaping bacterial metabolism and cluster architecture.
RESULTS AND DISCUSSION
Variations in isomerase and reductase clusters across bacterial genomes
There are 16 potential combinations (types A–H and A^c^–H^c^) of clusters encoding isomerases (KduI or DhuI) and reductases (KduD or DhuD), as illustrated in Fig. 3 and 4. Using representative sequences from 3,550 bacterial strains (Table S1), DHU isomerases and DK-II reductases were identified by BLAST and hidden Markov model (HMM) searches (28, 29). Among the 3,550 representative genomes, 568 clusters were identified, and 538 strains (15%) harbored putative clusters, indicating that some strains possess two or more clusters in their genomes. Among the 16 possible types of cluster combinations, 10 were identified in bacterial genomes (Fig. 3 and 4). As most of the clusters (562 of 568 clusters) arranged the two genes with the same direction (types A–H), we focused on all cluster types excluding C in the subsequent analysis. Among the seven investigated types, strains harboring the cluster kduI–kduD (type A) were most common (280 strains and 11 phyla; Tables S3 and S4), followed by those harboring kduI–dhuD (type E; 134 strains and seven phyla; Fig. 4B; Tables S3 and S7). The cluster kduD–kduI (type B), in which the order of the two genes is reversed from that in type A clusters, was widely distributed independent of the phylum and type A clusters (Tables S3 and S5). The cluster dhuD–dhuI (type D) was detected only in the phyla Bacillota (30 strains) and Actinomycetota (six strains; Tables S3 and S6). Moreover, 90% of strains possessing the hybrid cluster kduI–dhuD (type E) belonged to either Bacteroidota (63%) or Bacillota (27%; Fig. 4C; Table S3 and S7). The type F cluster dhuD–kduI was detected specifically in the phyla Mycoplasmatota and Spirochaetota (Fig. 4B; Tables S3 and S8). Strains harboring the hybrid dhuI–kduD cluster (type G) were limited to the families Sphingomonadaceae (nine strains) and Erythrobacteraceae (four strains) in the phylum Pseudomonadota (Fig. 4C; Tables S3 and S9). Type H clusters were primarily detected in the phylum Pseudomonadota, but they were also present in Faecalibacterium duncaniae (strain JCM 31915, phylum Bacillota). The cluster kduD–dhuI (type H) was detected in 41 strains, of which 40 belonged to the phylum Pseudomonadota (Tables S3 and S10). Approximately 18% of strains (243 of 1,378 strains) belonging to Pseudomonadota harbored clusters of any type, primarily types A (13.9%) and H (2.9%).
Possible configurations of isomerase–reductase clusters. Types A–H contain genes encoding isomerases and reductases oriented in the same direction. Types AC–HC represent clusters in which the two genes are arranged in opposite directions. The number of cluster-positive strains and phyla is presented to the right of each cluster diagram.
Pan-genome analysis of the cluster types. (A) A phylogenetic tree based on core genomes and cluster types (types A–H). The figure was drawn using iTOL (version 6) (30). (B) Proportions of phyla in each type. The numbers above the bar graphs represent the total number of strains harboring each cluster. (C) Proportions of cluster types in the phyla Pseudomonadota, Bacteroidota, and Bacillota.
Regarding DHU production, Hobbs et al. (11) reported that acquisition of kdgF provides an advantage by enabling enzymatic ring opening of unsaturated glucuronic acid. Among the 538 cluster-positive strains, the proportion of kdgF-positive strains was <50% for all types excluding type E-positive strains (Fig. S1A), indicating that in most strains, unsaturated glucuronic acids are converted to DHU nonenzymatically in the absence of KdgF.
Among type A–H clusters (562 detected clusters) , 552 (98%) and 552 (98%) clusters co-occurred with kdgA and kdgK, respectively, which encode the DHU-metabolizing enzymes 2-dehydro-3-deoxyphosphogluconate aldolase and 2-dehydro-3-deoxygluconokinase, respectively (Fig. S1B and Table S14), suggesting that the cluster-possessing strains metabolizes DHU after enzymatic reaction by isomerases and reductases.
Of the 3,550 strains analyzed, 3,012 lacked DHU metabolism–related gene clusters. Among these, kduI was detected in 201 strains, all of which carried kduD. dhuI was identified in 144 strains, of which 124 and 141 strains also harbored dhuD and kduD, respectively. This suggests that these strains retain isomerase and reductase genes at separate loci on the chromosome rather than as part of an operon or gene cluster. Additionally, 73 strains, including those from the genus Mycoplasma, lacked kduI, kduD, dhuI, and dhuD.
To explore whether isomerase and reductase genes are organized as clusters, we calculated the genomic distance between the two genes (Fig. 5), which revealed a close distance for all types, excluding type H (kduD–dhuI cluster). Of the 41 type H strains, 12 carried a gene encoding a protein with a cupin-domain protein located between kduD and dhuI. These proteins, characterized by the presence of a cupin domain, were identified as KdgF, which facilitates the transformation of cyclic unsaturated uronates into their linear ketone forms (29), opposed to KduI, which exhibits similarity to cupin-type phosphoglucose isomerase (31). Furthermore, in 13 of the 41 strains, a gene identified as kduI was located upstream of kduD, forming a tandem cluster kduI–kduD–dhuI, whereas the remaining 28 strains harbored simple type H clusters in the absence of kduI. These data suggested that type H clusters were formed through different evolutionary processes.
Distance between isomerase and reductase genes. (A) Bar plot of distance between genes and frequency with a cutoff of 1000 bp for each cluster (types A–H). (B) Box plots of distance for each cluster with a cutoff of 500 bp.
Insights into the phyla Bacteroidota and Bacillota
We further analyzed the diversity of clusters in the phyla Bacteroidota and Bacillota, in which strains bearing the type E hybrid cluster were abundant (Fig. 4C; Table S1). Bacteroidota contained type A, B, or H clusters, whereas one of the strains (Wenyingzhuangia fucanilytica strain CZ1127) harbored a tandem cluster composed of kduD–kduI–dhuD. Compared with that in other phyla, the possession ratio of type A clusters was low in Bacteroidota (17 of 309 strains). Bacillota contains the families Streptococcaceae (15 of 51 strains were cluster-positive), Clostridiaceae (14 of 41 strains were cluster-positive), and Lachnospiraceae (17 of 34 strains were cluster-positive), which are common residents in the oral cavities and guts of humans and animals. By contrast, no cluster-positive strains were detected within Staphylococcaceae, a family typically residing in the nasal cavity. Although Staphylococcus aureus secretes hyaluronate lyases, these lyases are involved in the dispersion of hyaluronic acid–containing biofilms and subsequent distribution of the pathogenic bacterium to host tissues, rather than the utilization of hyaluronic acid as a nutrient source (32). Streptococcaceae and Clostridiaceae harbored primarily type D (dhuD–dhuI) clusters. Five strains in the family Lachnospiraceae (Marvinbryantia formatexigens and four Blautia species) harbored the tandem kduI–dhuD–dhuI cluster.
Correlation with DHU-producing enzymes
Based on the observed habitat preferences, we hypothesized that the possession of GAG- and pectin-degrading enzymes could explain the patterns of environmental adaptation. Oligomerized GAGs are degraded by UGLs, whereas pectins are cleaved by OGLs and/or YteRs. Enzymatic reactions catalyzed by UGLs, OGLs, and YteRs commonly result in the production of DHU, which serves as the substrate of the isomerases KduI and DhuI. Therefore, the presence of UGLs, OGLs, and YteRs is suspected to be associated with the environmental adaptation of bacteria. We compared the rates of UGL, OGL, and YteR gene positivity among the seven types of clusters (Fig. S1C). Strains containing type B, D, E, or F clusters harbored UGL genes at high rates, indicating that the acquisition of these clusters is advantageous for colonization of human hosts. Although OGLs and YteRs target oligomerized pectin, the presence of clusters in the genome varied among type D-, E-, and F-positive strains.
M. formatexigens (DSM 14469), a human gut-commensal anaerobe bacterium, harbors kduI–dhuD–dhuI, which is positive for types D (dhuD–dhuI) and E (kduI–dhuD), as well as a UGL gene, whereas it lacks OGL and YteR genes. Although the UGL gene was detected in type D-positive strains, none of them harbored the OGL gene. However, a previous study demonstrated that this bacterium harbors 103 GHs, including GH28 family hydrolases (polygalacturonases), which are involved in pectin digestion (33), indicating that pectin is degraded into saturated galacturonic acid and metabolized independen tly of KduI or DhuI. In addition to M. formatexigens, the kduI–dhuD–dhuI cluster was detected in four Blautia species, namely, Blautia producta (DSM 2950), Blautia pseudococcoides (strain YL58), Blautia hansenii (DSM 20583), and Blautia argi (KCTC 15426). UGL genes were detected in three Blautia species (B. producta, B. pseudococcoides, and B. hansenii), and B. producta also harbored YteR, suggesting that these three species can metabolize GAGs. None of the Blautia species harbored an OGL gene. It remains unclear whether Blautia species utilize pectin as a nutrient. Dang et al. (34) reported that pectin supplementation to piglets increased the relative abundance of Blautia. Conversely, Larsen et al. (35) demonstrated that pectin supplementation decreased the relative abundance of Blautia. As changes in the relative abundance of Blautia do not reflect pectin content, further analysis is required to clarify pectin utilization by Blautia.
Prevalence of the clusters in human gut microbes
The family Bacteroidaceae, which contains the genera Bacteroides and Parabacteroides, harbored only the type E cluster kduI–dhuD, which is a hybrid type of kdu and dhu. Genus Bacteroides is abundant in the human gut, which is rich in mucus-derived GAGs and dietary pectins. Previous reports found that B. thetaiotaomicron and Bacteroides xylanisolvens harbor specific polysaccharide utilization loci that assimilate pectin with different degrees of esterification (36–38). Therefore, we hypothesized that the presence of type E clusters in the genome is advantageous in environments where both GAGs and pectins are abundant. To examine the hypothesis, we analyzed the relative abundance of the clusters in human feces using metataxonomic data from 290 healthy adults (Fig. 6). Consistent with our hypothesis, the relative abundance of type E clusters was significantly higher than that of other types (P < 0.0001 by the Kruskal–Wallis and Wilcoxon tests). Regarding the phylum Bacillota, type E clusters were detected in the families Lachnospiraceae (16/34 strains), Enterococcaceae (10/23 strains), and Streptococcaceae (3/51 strains). All of these type E-positive strains, excluding the two Lactococcus species in the family Streptococcaceae, have been reported as human gut-commensal bacteria (39, 40), supporting our hypothesis. Moreover, UGL genes were detected at the highest rate in type E-positive strains (Fig. S1). Although OGL genes were rarely detected in type E-positive strains, they harbored YteR genes at the highest rate, in addition to UGL genes (Fig. 6). These data indicate that the possession of UGL and YteR genes is advantageous for gut colonization, similar to type E clusters.
Relative abundance of the clusters in healthy adult guts. The relative abundance of types A–H and the enzyme-encoding genes (ugl, ogl, pelW, and YteR) were estimated using the metataxonomic data of 290 healthy adults in the United States.
Habitant preference
To explore whether the presence of clusters in the genome is associated with environmental adaptation, habitant preference was estimated using the ProkAtlas database (41). The average scores of habitant preference varied widely among clusters (Fig. 7; Fig. S3). Type A was commonly found in human gut, soil, and rhizosphere samples. Type D was frequently observed in the human gut but rarely detected in soil, rhizosphere, or marine environments. In contrast, type G was not detected in the human gut but tended to occur in marine, soil, and rhizosphere samples. Type E was frequently present in human gut and marine environments. Type H was broadly distributed and frequently found in the following order: annelid gut, rhizosphere, and human gut.
Heatmap of habitat preference scores for selected categories. Habitat preference scores were calculated using ProkAtlas (3, 41). Values inside the boxes indicate average habitant preference score. White boxes, scores of the overall average across all cluster types; red, scores above the average across all cluster types; purple, scores below the average across all cluster types. The categories (soil, rhizosphere, marine, human oral, human gut, gut, and annelid) were selected according to an average score >10 in at least one operon. Average habitat preference scores of all categories are presented in Supplementary Material (Fig. S3).
In the human gut, GAGs serve as major nutrient sources for the gut microbiota because they are richly present as free molecules and components of proteoglycans and they are continuously shed from the gut epithelium and mucosal layer (6). The marine environment is abundant in alginate, which is primarily produced by seaweeds (42). In soil and rhizosphere, pectin is supplied from plant cell walls, and its concentration is associated with biofilm formation and bacterial colonization (43). Our results indicated the association of clusters with environmental adaptation. Differences in substrate availability among environments might explain the distribution patterns of cluster types.
Phylogenetic analysis based on isomerase and reductase
We conducted a phylogenetic analysis based on the amino acid sequences of isomerases and reductases (Fig. 8). Although the phylogenetic tree of KduI was constructed with five clades (Fig. 8A), the clades were not correlated with cluster types, indicating that variations in KduI sequences occurred before cluster formation. By contrast, variations in DhuI were correlated with cluster types (clade 1, type D; clade 2, type G; and clade 3, type H; Fig. 8B). This correlation indicated that the variations in DhuI occurred through different selection pressures among cluster-possessing bacteria after cluster formation. DhuI in the phylum Actinomycetota did not form an independent clade, but it clustered with that of the phylum Bacillota similarly as DhuD (Fig. S1B). The similarity of DhuI and DhuD between the different phyla suggested horizontal gene transfer of the dhuD–dhuI cluster from Bacillota to Actinomycetota.
Phylogenetic tree of isomerases. (A) KduI. (B) DhuI. The tree was constructed using RaxML (42). The figure was drawn using iTOL (version 6) (30).
Limitations
This study had several limitations. Although we focused on DHU metabolism–related gene clusters, their presence alone does not fully explain adaptability to different environments. For instance, Browne et al. (44) reported that Firmicutes bacteria that lost sporulation capability were more abundant within individual hosts but less abundant than spore-forming Firmicutes bacteria. Similarly, Levy et al. (45) highlighted that multiple genomic traits contribute to plant-associated adaptation. As shown in Table S1, some bacteria duplicated reductase genes. In particular, Rhodococcus pseudokoreensis possesses 146 KduD genes, and Mycolicibacterium moriokaense possesses 50 DhuD genes. The large number of duplicate detections likely indicates a high rate of false positives in our gene identification approach. As homologous genes were identified using HMM and BLAST searches, it is possible that homologous proteins with different substrate specificities were incorrectly detected. As presented in Fig. 2, the isomerase and reductase genes were oriented in the same direction across types A–H clusters. In most cases, the distance between these two genes was <100 bp, except for type H (Fig. 5), suggesting that they might be conserved as clusters. However, further analysis is needed to determine whether the genes are indeed cotranscribed.
Conclusion
This pan-genome analysis found a wide distribution of type A clusters independent of the phylum, suggesting that the type A cluster kduI–kduD represents the ancestral form, which was subsequently replaced during the course of evolution. The hybrid cluster kduI–dhuD (type E) might play a role in bacterial colonization in the intestinal environment through the metabolism of GAGs and pectins by UGLs and YteRs, respectively. Moreover, some strains harbored a tandem arrangement of three genes. This study proposes a correlation between Kdu/Dhu genes involved in the assimilation of DHU and the evolution of bacterial persistence.
MATERIALS AND METHODS
Data sources
Representative and complete genomes that passed the taxonomy check and their annotated protein sequences were obtained from the National Center for Biotechnology Information RefSeq database (retrieved 13 January 2023) (46) (Table S1). The genomes of 3,550 strains were collected, including two genomes derived from Escherichia coli (O157:H7 strain Sakai and strain K-12 substrain MG1655). Table S1 lists the accession numbers of the genomes. The genomes were classified using TaxonKit (version 0.17.0) (47). As listed in Table S3, 1,378, 653, 641, and 309 strains belonging to the phyla Pseudomonadota, Actinomycetota, Bacillota, and Bacteroidota, respectively, were included in this study.
Phylogenetic analysis
The core genes in the 3,550 complete genomes were detected using the UBCG2 pipeline (48). Based on the concatenated sequences of the core genes, a phylogenetic tree was constructed using FastTree (49). Data sets of Bacteroidota and Bacillota were extracted to construct rooted phylogenetic trees with Chlorobium limicola DSM 245 and Thermotoga maritima strain MSB8 for the outgroups of Bacteroidota and Bacillota, respectively. Protein sequences (KduI, KduD, DhuI, and DhuD) were aligned using MAFFT (version 7.525) (50), followed by trimming of poorly aligned regions using the “automated1” method in trimAl (version 1.5.0) (51). The trimmed alignment was used as input for the phylogenetic analysis using RAxML (version 8.2.13) with the PROTGAMMALG model (52). The phylogenetic trees were visualized using iTOL (version 6) (30).
Homologous gene search based on the HMM profile
To generate the HMM profile, 20 nonredundant sequences were selected as described by Bundalovic-Torma et al. (53) with some modifications. As initial queries, the amino acid sequences of L. rhamnosus strain Lc705 KduI (CAR91530.1), L. rhamnosus strain Lc705 KduD (CAR91531.1), S. agalactiae strain NEM316 DhuI (CAD47551.1), S. agalactiae strain NEM316 DhuD (CAD47550.1), S. agalactiae strain NEM316 UGL (CAD47548.1), D. dadantii strain 3937 OGL (ADM98639.1), Bacillus subtilis subsp. subtilis strain 168 YteR (unsaturated rhamnogalacturonyl hydrolase of the GH105 family) (CAB14990.1), L. rhamnosus strain Lc705 KdgA (2-dehydro-3-deoxyphosphogluconate aldolase/4-hydroxy-2-oxoglutarate aldolase) (CAR91532.1), L. rhamnosus strain Lc705 KdgK (2-dehydro-3-deoxygluconokinase) (CAR91533.1), and Y. enterocolitica subsp. enterocolitica strain 8081 KdgF (pectin degradation protein) (CAL11968.1) were selected (Table S2). The protein BLAST search was conducted using the annotated proteins from the 3,550 genomes as references with a threshold of E < 1 × 10^−5^ (28). The detected homologous sequences were collected as data sets. Among all detected homologous sequences, those with <97% sequence identity and with the lowest E value were selected as the next queries. The sequences of the first queries were removed from the data sets. Then, using the next queries and data sets, a protein BLAST search was conducted to detect new queries. By repeating these procedures 20 times, we obtained 21 query sequences for each protein. These selected query sequences were aligned using MAFFT (version 7.525) (50). An HMM profile was generated from the aligned sequences using HMMER (version 3.4) (54). Based on the generated profiles, homology searches were conducted using the complete genomes of 3,550 strains. Finally, genes whose complete sequences had E < 1 × 10^−5^ were considered homologous.
Identification and classification of GAG metabolism–related gene clusters
To determine whether GAG isomerase and reductase are organized in a cluster, we calculated the distance between isomerase (kduI or dhuI) and reductase (kduD or dhuD) genes detected using the aforementioned HMMER search. If the distance between the gene pairs was ≤500 bp, then the pairs were defined as putative clusters. According to the types of genes and order, the putative clusters were classified into ten types (Fig. 2; Table S4 through S10).
Fecal microbiomes of healthy adults
Metagenomic data from the fecal microbiomes of 290 healthy US adults (BioProject accession number: PRJNA795985) were analyzed in this study. Paired-end 151-bp raw sequencing reads (Run IDs, SRR17531755–SRR17532044) were retrieved using the SRA Toolkit (https://hpc.nih.gov/apps/sratoolkit.html). Quality filtering and adapter trimming were performed using fastp (55). To remove host-derived reads, sequences mapped to the human reference genome (GRCh38/hg38), which was downloaded from the UCSC Genome Browser (56), were filtered using Bowtie2 (57) and SAMtools (58). A bacterial genome database was constructed using 3,550 representative bacterial genomes, as listed in Table S1, and indexed with Kraken2 (59). Cleaned reads were classified by Kraken2, and abundance estimates were generated using Bracken (60).
Habitat preference score estimation
rRNA gene sequences were detected using barrnap (https://github.com/tseemann/barrnap). 16S rRNA gene sequences were extracted using seqkit (61). After clustering the 16S rRNA gene sequences with a threshold of 90% sequence identity, a centroid sequence of the largest cluster was obtained using VSEARCH (62) and used as the representative 16S rRNA gene sequence. When two or more clusters contained the largest number of sequences, the longer/longest centroid sequence was used. The habitat preference score of each bacterium was calculated using ProkAtlas with default parameters based on the representative 16S rRNA gene sequences (41). Using gene cluster information listed in Table S1 (A–H, A^C^–H^C^), the average scores of habitat preference are presented in Fig. 7.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Casale J, Crane JS. 2023. Biochemistry, glycosaminoglycans. Stat Pearls Publishing.31335015 · pubmed ↗
- 2Song Y, Zhang F, Linhardt RJ. 2021. Glycosaminoglycans. Adv Exp Med Biol 1325:103–116. doi:10.1007/978-3-030-70115-4_434495531 · doi ↗ · pubmed ↗
- 3Kamhi E, Joo EJ, Dordick JS, Linhardt RJ. 2013. Glycosaminoglycans in infectious disease. Biol Rev Camb Philos Soc 88:928–943. doi:10.1111/brv.1203423551941 · doi ↗ · pubmed ↗
- 4Marion C, Stewart JM, Tazi MF, Burnaugh AM, Linke CM, Woodiga SA, King SJ. 2012. Streptococcus pneumoniae can utilize multiple sources of hyaluronic acid for growth. Infect Immun 80:1390–1398. doi:10.1128/IAI.05756-1122311922 PMC 3318431 · doi ↗ · pubmed ↗
- 5Nguyen VA, Ogura K, Matsue M, Takemoto N, Mukai K, Nakajima Y, Hoang TL, Iwata Y, Sakai N, Wada T, Hashimoto W, Okamoto S, Ichimura H. 2020. Novel hyaluronate lyase involved in pathogenicity of Streptococcus dysgalactiae subsp. equisimilis. Front Microbiol 11:552418. doi:10.3389/fmicb.2020.55241833072013 PMC 7541959 · doi ↗ · pubmed ↗
- 6Rawat PS, Seyed Hameed AS, Meng X, Liu W. 2022. Utilization of glycosaminoglycans by the human gut microbiota: participating bacteria and their enzymatic machineries. Gut Microbes 14:2068367. doi:10.1080/19490976.2022.206836735482895 PMC 9067506 · doi ↗ · pubmed ↗
- 7Dong J, Cui Y, Qu X. 2024. Metabolism mechanism of glycosaminoglycans by the gut microbiota: bacteroides and lactic acid bacteria: a review. Carbohydr Polym 332:121905. doi:10.1016/j.carbpol.2024.12190538431412 · doi ↗ · pubmed ↗
- 8Oiki S, Nakamichi Y, Maruyama Y, Mikami B, Murata K, Hashimoto W. 2019. Streptococcal phosphotransferase system imports unsaturated hyaluronan disaccharide derived from host extracellular matrices. P Lo S One 14:e 0224753. doi:10.1371/journal.pone.022475331697725 PMC 6837340 · doi ↗ · pubmed ↗