The Multilayered Landscape of Ferroptosis: Plasticity, Propagation, and Evolutionary Perspectives
Hong Chen, Hongfa Yan, Hong Bu, Feng Ye

TL;DR
This paper explores ferroptosis, a type of cell death, focusing on its flexible mechanisms, how it spreads, and its evolutionary significance.
Contribution
The paper provides a comprehensive framework highlighting the plasticity and propagation of ferroptosis across biological systems.
Findings
Ferroptosis involves flexible lipid peroxidation and antioxidant systems rather than a fixed pathway.
Ferroptosis can propagate through intracellular and intercellular mechanisms, leading to tissue damage.
Ferroptosis-like processes are evolutionarily conserved and relevant in development and disease.
Abstract
Ferroptosis is a distinct form of regulated necrotic cell death driven by iron-dependent phospholipid peroxidation, characterized by flexible and context-dependent mechanisms rather than a single fixed linear pathway. This study elucidates the critical lipid peroxidation networks and antioxidant defense systems used in determining ferroptosis, specifically emphasizing how these mechanisms underpin the plasticity of this cell death mode and its correlation with therapeutic resistance. We examine the catastrophic propagation of ferroptosis, detailing the multi-layered amplification mechanisms—ranging from intracellular organelle crosstalk to intercellular trigger waves—that may facilitate massive tissue damage in degenerative diseases and ischemic injuries. Furthermore, the evolutionary conservation of ferroptosis-like phenomena across diverse species is summarized, underscoring its…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
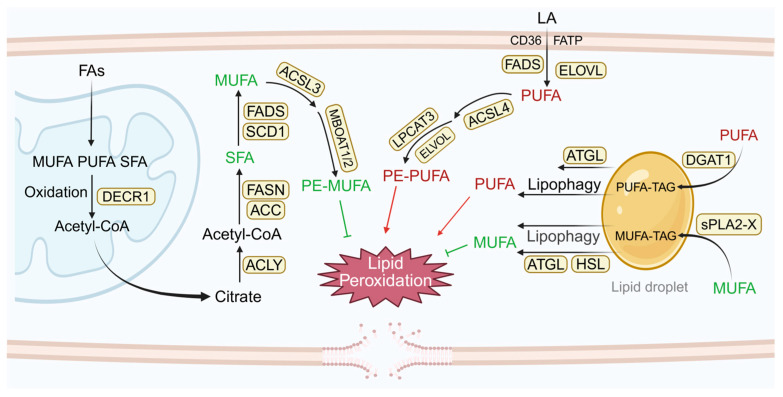 Figure 1
Figure 1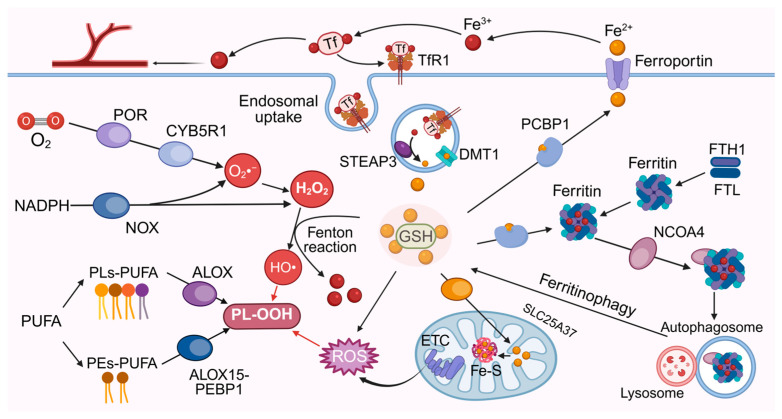 Figure 2
Figure 2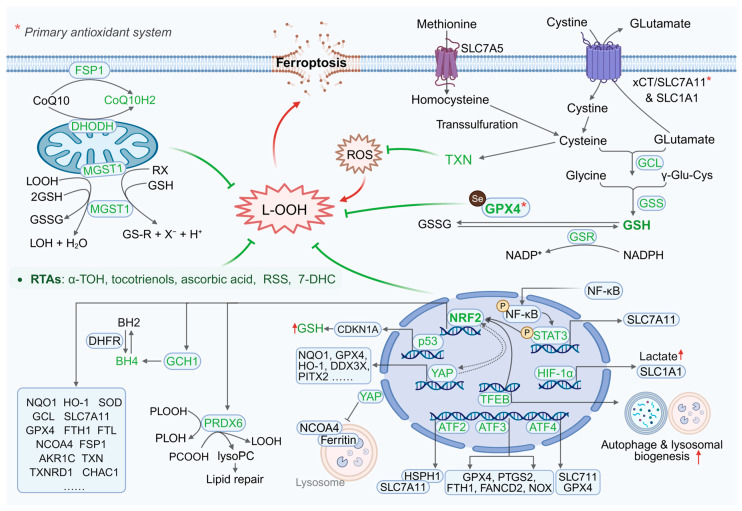 Figure 3
Figure 3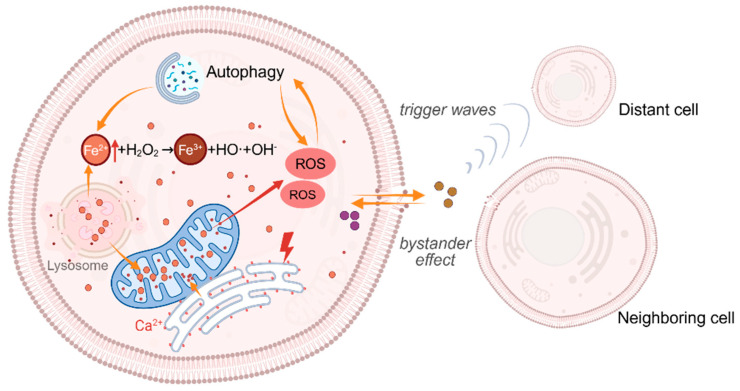 Figure 4
Figure 4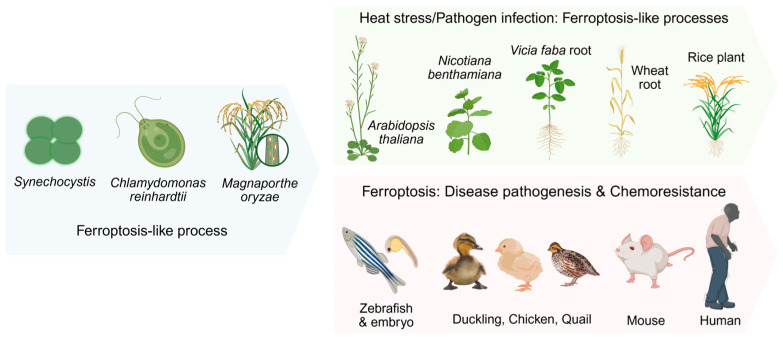 Figure 5
Figure 5- —Postdoctor Research Fund of West China Hospital, Sichuan University
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsFerroptosis and cancer prognosis · Immune cells in cancer · Clusterin in disease pathology
1. Introduction
Ferroptosis is a distinct form of regulated necrotic cell death characterized by iron-dependent phospholipid peroxidation, whose susceptibility is conserved and modulated by dysregulated iron metabolism and redox imbalance [1,2]. Accumulating evidence indicates that ferroptosis does not operate through a single obligatory molecular trigger, but instead emerges from the integration of multiple biochemical processes that vary across cellular and pathological contexts.
Two key observations exist in existing ferroptosis research: (1) although numerous metabolites and proteins can initiate or regulate ferroptosis, no single one is absolutely essential in all biological contexts; and (2) the same molecules can exhibit distinct pro- and anti-ferroptotic activities under different cell-type-specific signaling pathways and stress-dependent induction patterns. This operational paradigm requires defining ferroptosis as a death mode orchestrated by a combination of biochemical mechanisms in specific environments, rather than a strictly linear pathway. At the macromolecular level, ferroptosis fundamentally requires the catastrophic accumulation of lipid peroxides—a process that depends on three key elements: an oxidizable lipid substrate, molecular oxygen, and reactive iron to facilitate autocatalytic propagation through both enzymatic and non-enzymatic mechanisms [3]. These elements act as metabolic hubs, connecting interconnected networks encompassing lipid metabolism, iron homeostasis, antioxidant defense, and bioenergetics. The inherent complexity of these networks, coupled with tissue-specific regulatory pathways, endows ferroptosis with remarkable plasticity.
Beyond the single-cell level, ferroptosis has been increasingly associated with large-scale, progressive tissue injury and degenerative diseases [4,5,6], suggesting that ferroptosis undergoes a series of intracellular and extracellular amplification and diffusion events to support its feedback amplification. Understanding these feedback amplification mechanisms has the potential to prevent the progression of neurodegenerative diseases or induce ferroptosis in refractory cancers.
Importantly, lipid peroxidation itself is a fundamental biochemical process observed across a wide spectrum of species, ranging from plants and invertebrates to mammals, reflecting its deep evolutionary conservation in cellular physiology [7]. Because ferroptosis is defined by iron-dependent phospholipid peroxidation, this shared vulnerability of membrane lipids to oxidative damage raises the possibility that core features of ferroptosis are conserved across diverse biological systems.
This review will reveal the mechanistic plasticity and propagation characteristics of ferroptosis by dissecting the complex intracellular lipid peroxidation and antioxidant networks, and the spatiotemporally coordinated feedback and diffusion pathways, thereby advancing our understanding of ferroptosis. It will also summarize ferroptosis-like phenomena in various species, emphasizing their potential physiological relevance and evolutionary significance.
2. Mechanistic Plasticity: A Game Between Lipid Peroxidation and Antioxidant Networks
Cells typically possess multiple parallel signaling pathways to execute critical functions, ensuring robust survival. Many metabolites and proteins can initiate or regulate ferroptosis, yet not all are essential or sufficient within organisms. For example, acyl-CoA synthetase long-chain family member 4 (ACSL4) is widely considered as necessary for ferroptosis, and its mediated biosynthesis of polyunsaturated fatty acid (PUFA)-containing phosphatidylethanolamines (PEs-PUFA), which serve as preferential substrates for lipid peroxidation [8]. However, photodynamic therapy induces ferroptosis by directly peroxidizing PUFAs and generating exogenous reactive oxygen species (ROS) in an ACSL4-independent manner [9]. This phenomenon reveals profound redundancy and compensation mechanisms in cellular ferroptosis pathways. In addition, the same molecule may exhibit opposite regulatory effects on ferroptosis in different disease contexts and metabolic states. For instance, low-dose doxorubicin or short-term Nutlin treatment can induce p53 transactivation of calcium-independent phospholipase A_2_β (iPLA_2_β), thereby suppressing ferroptosis in melanoma and sarcoma cells [10]. In contrast, the earlier reports demonstrated that p53 can suppress solute carrier family 7 member 11 (SLC7A11) transcription, reduce glutathione levels and glutathione peroxidase 4 (GPX4) activity, and promote ferroptosis [11]. Recent genome-wide CRISPR–Cas9 screens identified 7-dehydrocholesterol (7-DHC) as an endogenous lipid factor that confers resistance to ferroptosis [12,13], Owing to a conjugated diene within its sterol core, 7-DHC efficiently scavenges lipid-derived radicals and terminates lipid peroxidation chain reactions. Functionally, 7-DHC acts as a sacrificial lipid antioxidant, preferentially undergoing oxidation to limit the propagation of oxidative damage to surrounding phospholipids, including ferroptosis-relevant PUFA-containing species. This mechanism highlights that lipid composition and intrinsic radical-trapping capacity modulate ferroptosis susceptibility. Together, these phenomena point to a core conclusion: ferroptosis is not a straightforward process controlled by a single linear pathway, but rather a highly dynamic, interconnected, and context-specific network of cell fate decisions.
2.1. Plasticity of Lipid Peroxidation
2.1.1. Diverse Substrates for Lipid Peroxidation
The initiation of both enzymatic lipid peroxidation and autoxidation relies on sufficient “fuel” reserves within the plasma membrane. Experimental evidence has shown that deuterium substitution at bis-allylic positions effectively suppressed autoxidation, validating the critical role of PUFAs in this process [14]. Notably, oxidation of esterified PUFAs is a prerequisite for classical ferroptosis mechanisms, particularly PEs-PUFA [15,16]. The ACSL4-lysophosphatidylcholine acyltransferase 3 (LPCAT3) axis orchestrates PE-PUFA synthesis and represents the main pathway in GPX4 inhibition-induced ferroptosis [17]. However, its importance is diminishes in models such as cystine deprivation [18], p53 induction [19], and photodynamic therapy [20]. Beyond PEs-PUFA, other phospholipids including phosphatidylcholine, phosphatidylserine, and phosphatidylglycerol can also promote ferroptosis [21]. The differential oxidizability among various phospholipids arises because the headgroup influences membrane localization, chemical properties, and membrane conformation [22]. Moreover, Qiu et al. [21] demonstrated that diacyl-PUFA-containing phosphatidylcholine exhibit even greater potency in promoting ferroptosis compared to PEs-PUFA supplementation. In addition to the phospholipids mentioned, peroxisome-derived polyunsaturated ether phospholipids can also participate in lipid peroxidation and drive ferroptosis [23]. However, polyunsaturated ether phospholipids are not inherently more susceptible to peroxidation than polyunsaturated phospholipids, and it remains unclear whether they can functionally replace polyunsaturated phospholipids in promoting ferroptosis.
In contrast, the incorporation of oxidation-resistant monounsaturated fatty acids (MUFAs) into membrane phospholipids protects cells from ferroptosis. MUFAs are competitively incorporated into phospholipids through ACSL3-membrane-bound O-acyltransferase domain-containing 1 and 2 (MBOAT1/2), reducing the PUFA/MUFA ratio and thus inhibiting lipid peroxidation [24]. Lipid droplets (LDs) are storage organelles composed mainly of triglycerides and sterol esters. Nutritional and oxidative stressors modulate LD biogenesis to regulate membrane unsaturation through dynamic PUFA/MUFA partitioning between triglycerides and phospholipid pools, thereby influencing ferroptosis susceptibility [25]. The diacylglycerol acyltransferase 1 (DGAT1)-mediated PUFA channeling into LDs protects against oxidative stress-induced cell death under exogenous PUFA overload [26]. Conversely, LDs may also promote ferroptosis through either direct peroxidation or context-dependent release of PUFAs [27,28] (Figure 1).
Therefore, ferroptosis does not rely on a single lipid class or a fixed pathway. Instead, its execution mechanism adapts dynamically according to the substrate availability, abundance, and the cellular environment.
2.1.2. Redundancy in Lipid Peroxidation Triggering Pathways
The lipid peroxidation process consists of two phases: initiation (radical generation) and amplification (chain reaction). The initiation can proceed through multiple parallel routes: the non-enzymatic pathway generates ROS through the Fenton reaction mediated by ferrous ions (Fe^2+^) or mitochondrial electron leakage, which initiates lipid radicals and triggers an autocatalytic cascade reaction, constituting the core amplification mechanism of lethal accumulation of lipid peroxides [2,29]; In parallel, enzymatic pathways provide diverse initiation mechanisms, including oxidation of specific phospholipid substrates by arachidonate lipoxygenase (ALOX) family [30], NADPH-dependent hydrogen peroxide production by oxidoreductase NADPH-cytochrome P450 reductase (POR)/NADH-cytochrome b5 reductase (CYB5R1) [31], and tissue-specific ROS generation by NAD(P)H oxidase (NOX) family [32] (Figure 2). Enzymatic lipid peroxidation may predominantly function during the initiation phase, while subsequent amplification largely relies on non-enzymatic autoxidation. For example, in type II ferroptosis inducer (RSL3)-induced ferroptosis, intracellular peroxidized lipids surge within 1 h of treatment, culminating in cell death within 4 h [18]. If enzymatic processes dominated this rapid progression, such dramatic lipid peroxidation kinetics would require concurrent upregulation of peroxidation-related enzymes, but this phenomenon not supported by empirical evidence.
Different triggers may activate distinct dominant pathways to induce ferroptosis. In the R6/1 Huntington’s disease model, loss of ALOX5 expression eliminates HTTQ94-associated ferroptosis under ROS or glutamate stress and ameliorates pathological phenotypes [33]. Meanwhile, NOX2 plays a critical role in stress-induced ferroptosis in retinal ganglion cells [34]. Similarly, different enzymatic pathways predominate in different cell types. Inhibition of ALOX5 alleviates dopaminergic neuronal ferroptosis and motor deficits in a Parkinson’s disease mouse model [35], and its protective role extends to mouse models of stroke [36] and epilepsy [37]. On the other hand, the NOX family may play a more prominent role in tumor contexts. For instance, NOX2 and NOX4 enhance ferroptosis in epithelial ovarian cancer and renal malignancies, respectively [38,39]. Notably, significant functional redundancy exists among these enzymatic pathways, as evidenced by the fact that the loss of ALOX can be compensated for by POR or non-enzymatic mechanisms [2,40,41].
Additionally, iron bioavailability is a key determinant of ferroptosis. As an essential enzymatic cofactor and catalyst in Fenton reactions, iron dynamically integrates the efficiency of various pathways through networks involving transferrin receptor 1 (TfR1)-mediated endocytosis, ferritin storage, and nuclear receptor coactivator 4 (NCOA4)-mediated ferritinophagy [42]. Consequently, the iron regulatory network, encompassing uptake, storage, export, and intracellular utilization, critically governs ferroptosis dynamics [42] (Figure 2). This multi-tiered regulatory system underpins the remarkable plasticity of ferroptosis. It enables the sustained maintenance of lipid peroxidation through the release of stored iron from ferritin, even in the face of impaired iron uptake, thereby guaranteeing the adaptive propagation of death signals across diverse stress conditions.
2.2. Plasticity of the Antioxidant Defense System
2.2.1. Core Defense Systems and Compensatory Mechanisms
To cope with oxidative damage, cells possess a robust and multi-layered antioxidant defense system (Figure 3). Within this system, GPX4 serves as the central defense against ferroptosis by uniquely catalyzing the reduction in phospholipid hydroperoxides [43]. The loss or inhibition of GPX4 can be partially compensated for by parallel antioxidant mechanisms [44]. Membrane-localized ferroptosis suppressor protein 1 (FSP1) utilizes NAD(P)H to reduce ubiquinone to ubiquinol (CoQH_2_), acting as a lipophilic radical-trapping antioxidant (RTA) to directly quench lipid radicals [45]; peroxiredoxin 6 (PRDX6) scavenges short-chain hydroperoxides through its peroxidase activity and hydrolyzes phospholipid hydroperoxides to generate lysophospholipids through phospholipase A_2_ (PLA_2_) activity, thereby synergistically limiting membrane damage propagation [46]; metabolic networks such as the GTP cyclohydroxylase 1 (GCH1)-tetrahydrobiopterin (BH4) axis neutralize lipid peroxyl radicals via BH4 [47], while enzymes like aldo-keto reductase family 1 member C1 (AKR1C1) detoxify cytotoxic aldehyde byproducts derived from lipid peroxidation [48]. Although these systems do not directly reduce phospholipid hydroperoxide, they dynamically establish compensatory defenses upon GPX4 dysfunction by limiting radical accumulation, removing oxidized lipid species, and detoxifying secondary metabolites. This “core is irreplaceable—bypass pathways are compensatory” architecture confers significant environmental adaptability to ferroptosis regulation, allowing cells to selectively mobilize dominant antioxidant routes to maintain redox homeostasis.
2.2.2. Diversity of Antioxidant Molecules
Some endogenous molecules can provide rapid protection by directly quenching lipid radicals or interrupting peroxidation chain reactions [49]. For instance, vitamin E (e.g., α-tocopherol) [50] and reduced coenzyme Q10 (CoQ10H_2_) [51,52] directly quench lipid peroxyl radicals through hydrogen atom transfer, anchoring within biological membranes to provide immediate protection. Beyond vitamin E, other vitamins have also been reported to have antioxidant effects [53]. Vitamin K forms (specifically menaquinone-4) exhibit potent radical-neutralizing capabilities [54].Vitamins A and D primarily modulate antioxidant gene expression via binding to specific nuclear receptors, indirectly conferring protection [55,56,57,58]. In addition, ascorbic acid (also known as vitamin C) can prevent ferroptosis through the nuclear factor erythroid 2-related factor 2 (NRF2) and SLC7A11-GPX4 pathways [59,60]. H_2_S-derived reactive sulfur species (RSS) can diffuse intracellularly to scavenge a variety of radicals by virtue of their strong reducing capacity [61]. Recent reports have also found that the presence of 7-DHC with a B-ring diene structure is a highly efficient RTA on the mitochondrial membrane [12,13]. Additionally, even molecules present at low concentrations, such as BH4, contribute by efficiently quenching superoxide and lipid radicals [62].
These molecules enable spatial complementarity within the antioxidant defense network. Specifically, vitamin E and CoQ10H_2_ synergistically protect the plasma membrane [63]; vitamin A-related retinoids contribute both direct antioxidant buffering at membranes and longer-term transcriptional reinforcement of ferroptosis defense programs [58]; 7-DHC safeguards mitochondrial and other cellular membranes [12], and RSS scavenges radicals within the cytosol [64]. When subjected to specific stressors, cells mobilize the most appropriate molecules to mount a primary defense and elicit synergistic protection. Vitamin E/CoQ10H_2_ help maintain basal redox homeostasis, while RSS offers broad scavenging capacity during ROS bursts. Furthermore, retinoids integrate redox control with lipid remodeling and the induction of ferroptosis-resistance gene, while 7-DHC is activated in response to mitochondrial stress. Therefore, cells flexibly call on optimized defense strategy according to the intensity of oxidative stress, subcellular damage sites, and metabolic status, highlighting the dynamic adaptability of the ferroptosis defense network.
2.2.3. Transcriptional Remodeling of Antioxidant Defenses
Transcriptional regulators confer plasticity in ferroptosis resistance by dynamically modulating antioxidant defense pathways. As a master regulator, NRF2 maintains redox and iron metabolism homeostasis by upregulating antioxidant enzymes (e.g., GPX4, SLC7A11, FSP1) [65,66] and lipid peroxidation inhibitors (e.g., AKR family members, PPARG) [67,68] (Figure 3). Its targeted inhibitors plumbagin or ML385 can enhance ferroptosis sensitivity in cancer therapy [69,70,71,72], while activators salidroside or sulforaphane play a protective role in neurodegenerative and metabolic diseases [73,74]. The activating transcription factor (ATF) family members (ATF1-4) regulate cell ferroptosis in a context-dependent manner by recognizing and binding to cAMP response elements (CREs) and influencing the transcription of multiple target genes [75]. Of which, ATF2 enhances antioxidant defense by stabilizing SLC7A11 [76], ATF3 inhibit SLC7A11/GPX4 to promote ferroptosis [77,78], and can also be regulated by NRF2 to enhance glutathione protection [79]. ATF4 inhibits lipid peroxidation by inducing the heat shock protein family A member 5 (HSPA5)-GPX4 axis [80], upregulating SLC7A11 [81], or activating the nuclear protein 1 (NUPR1)-lipocalin 2 (LCN2) pathway [82], and cooperates with NRF2 and YAP/TAZ to maintain redox homeostasis [83,84]. As a member of the nuclear factor of κ-light chain of enhancer-activated B cells (NF-κB) family, NF-κB p65 suppresses ferroptosis by directly regulating genes related to iron accumulation and/or lipid peroxidation [85]. Specifically, it transactivates SLC7A11 [86], induces LCN2 [87] and upregulates antioxidant proteins thioredoxin, heme oxygenase 1 (HO-1), and ferritin heavy chain 1 (FTH1) [88], and its phosphorylation level is associated with chemotherapy resistance [87]. Similarly to ATF3, p53 also plays a dual role in regulating ferroptosis. It promotes ferroptosis by inhibiting stearoyl-CoA desaturase-1 (SCD1) [89] and MBOAT1 [90] or upregulating ACSL4 [91], yet also enhances antioxidant defense via phospholipid transfer protein (PLTP)-mediated PUFA sequestration [92], induction of cyclin-dependent kinase inhibitor 1A (CDKN1A) expression [93], and suppression of the mevalonate pathway [94,95]. Furthermore, other factors can also modulate ferroptosis sensitivity to varying extents by remodeling the antioxidant network. YAP functions by cooperating with NRF2 [96], upregulating GPX4 [97], and blocking ferritinophagy [98]. Signal transducer and activator of transcription 3 (STAT3), through inducing SLC7A11 and promoting the incorporation of PUFAs into triglycerides, also plays a role [99,100]. The transcription factor EB (TFEB) contributes by activating the autophagy-NRF2 axis and maintaining iron homeostasis [101,102]. Additionally, hypoxia inducible factor-1 alpha (HIF-1α) enhances antioxidant genes via the NF-κB/Wnt pathway, further influencing ferroptosis regulation [103].
In summary, transcriptional factors underpin the plasticity of ferroptosis regulation via multi-layered control of key antioxidant and lipid peroxidation nodes. Their functions are highly dependent on disease context, microenvironment, and stress conditions. Precisely targeting specific transcriptional factors may offer novel therapeutic strategies for cancer and degenerative diseases.
3. Propagation: The Self-Catalyzing Storm of Lipid Peroxidation
Previous study has indicated a connection between ferroptosis and degenerative diseases characterized by extensive contiguous tissue damage [4]. As early as 2014, research by Linkermann et al. [104] demonstrated that, unlike control mice which all died within 48 to 72 h following 40 min of ischemia prior to reperfusion, Fer-1-treated mice did not develop functional acute renal failure or structural organ damage after ischemia–reperfusion injury (IRI). This suggests that ferroptosis mediates rapid and large-scale tubular cell death in renal IRI. Widespread contiguous spread of excessive cell death has also been observed across tissues in other models of IRI and degenerative disorders [105,106], implying that ferroptosis may can be amplified by a series of feedback/amplification loops that enable the rapid and sustained propagation of cell death events.
3.1. Intracellular: Multi-Layered Propagation Mechanisms
3.1.1. Radical Chain Reaction
The core propagation mechanism of ferroptosis originates from the radical chain reaction in non-enzymatic lipid peroxidation. The process is initiated when Fe^2+^ converts hydrogen peroxide into hydroxyl radicals via the Fenton reaction or when hydroxyl radicals are generated from superoxide dismutation derived from mitochondrial electron leakage [2,29,107]. In addition to these redox-driven routes, ferroptosis initiation can also occur through oxidase-mediated mechanisms, in which multiple oxidoreductases actively generate reactive oxygen species to initiate lipid peroxidation prior to extensive radical chain propagation [108]. Hydroxyl radicals selectively abstract the diallyl hydrogen atoms of PUFAs, forming lipid radicals. Lipid radicals rapidly react with molecular oxygen to generate highly reactive lipid peroxyl radicals, which propagate the chain by abstracting hydrogen from adjacent PUFAs, generating lipid hydroperoxides and regenerating lipid radicals. Lipid hydroperoxides further react with Fe^2+^ to generate lipid alkoxyl radicals, which rearrange to form epoxy peroxide radicals and regenerate peroxyl radicals, triggering a secondary peroxidation cascade [109]. The inherent instability of lipid hydroperoxides, peroxyl radicals, and lipid alkoxyl radicals lead to continuous production of oxidative products. Through a cyclic regeneration process, a self-sustaining feedback loop is established, resulting in exponential accumulation of lipid peroxides [30]. The intrinsic property of the radical reaction provides the fundamental driving force for the propagation nature of ferroptosis.
3.1.2. Autophagy-Driven Propagation of Ferroptosis
Growing evidence indicates that autophagy plays an important regulatory role in ferroptosis. This is supported by the observation of increased autophagy in erastin-induced mouse embryonic fibroblasts and HT1080 cells [110]. The fact that N-acetylcysteine treatment can block the induction of autophagy during ferroptosis, suggesting that autophagy may be triggered by high levels of intracellular ROS [110]. Additional evidence also shows that ROS accumulated during the early stages of ferroptosis activate pathways such as ataxia-telangiectasia mutated (ATM) kinase (a DNA damage response transducer) [111], mitogen-activated protein kinase (MAPK) [112], and the mechanistic target of rapamycin (mTOR) [113], which promote the expression of autophagy-related genes and initiate autophagic flux. Crucially, activated autophagy can further exacerbate lipid peroxidation through multiple effector pathways. For example, NCOA4-mediated ferritinophagy releases stored iron [114], while lipophagy degrades LDs to supply PUFA substrates [115,116]. Consistent with this, pharmacological inhibition of autophagy (BafA1/chloroquine) can reduce erastin/FIN56-induced ferroptosis and attenuates ROS increases triggered by ferroptosis inducers [117,118]. Thus, autophagy acts as a key bridging mechanism in the propagation of ferroptosis, forming a self-catalytic cycle of ROS increase and autophagy activation (Figure 4).
3.1.3. Inter-Organellar Crosstalk and Cascading Amplification
During ferroptosis, a series of physiological and biochemical reactions occur in the cell. These take place both in the cytosol and within organelles of the endomembrane system, including mitochondria, endoplasmic reticulum (ER), lysosomes, Golgi apparatus and peroxisomes. Alterations in the structure or function of these organelles, along with inter-organellar crosstalk, can synergistically promote the amplification of damage or death signals.
ER-mitochondria crosstalk constitutes an early driving hub in ferroptosis. Study has shown that ferroptosis inducers (e.g., RSL3) can trigger the dynamic remodeling of ER-mitochondrial contact sites (EMCSs) within minutes, including a significant expansion of the contact area and a shortening of the membrane distance [119]. This remodeling allows oxidized phospholipids (especially oxidized phosphatidylethanolamine and phosphatidylcholine) generated at this interface to be rapidly transferred to the mitochondrial membrane, leading to mitochondrial ROS generation and dysfunction [119]. Meanwhile, ROS derived from the mitochondrial respiratory chain can disrupt Ca^2+^ homeostasis in the ER, thereby hindering protein synthesis and release [120]. A large number of unfolded and misfolded proteins accumulate and cause ER stress. The ER subsequently releases Ca^2+^ into the mitochondria and cytoplasm, causing mitochondrial Ca^2+^ overload and further ROS production, forming an enhanced loop (Figure 4). In this process, proteins such as mitofusin 2 (MFN2) and its splice variant ERMIT2 regulate EMCS stability [121], while optic atrophy 1 (OPA1) affects the efficiency of ROS generation by maintaining the mitochondrial cristae structure and regulating the spatial structure of EMRCs [122], collectively determining the magnitude of cascade amplification.
Unlike the ER-mitochondria crosstalk, lysosome-mitochondria crosstalk provides a critical axis for iron metabolism amplification in ferroptosis (Figure 4). Lysosomes accumulate iron ions through the autophagic degradation of iron-rich proteins and cellular debris [123]. Increased lysosomal membrane permeability (e.g., after 4 h of RSL3 treatment) leads to rapid leakage of Fe^2+^ into the cytosol, directly catalyzing the Fenton reaction and driving lipid peroxidation [124]. Additionally, a “kiss and run” mechanism supports direct Fe^2+^ transfer from lysosomes to mitochondria [125]. This process involves membrane contact sites [126] and Fe^2+^ transport mediated by the divalent metal transporter 1 (DMT1)/the voltage-dependent anion channel 1 (VDAC1) located on the lysosomal and mitochondrial outer membranes [127]. Subsequently, mitoferrin2 on the mitochondrial inner membrane efficiently transports Fe^2+^ into the mitochondrial matrix, providing substrate for ROS burst and promoting the generation of toxic lipid radicals [128]. Furthermore, impaired mitochondrial function adversely affects lysosomal activity and acidification [129]. More critically, mitochondrial ROS can further elevate cellular iron levels via the autophagy-lysosome pathway, thereby activating ferroptosis [130]. Thus, iron trafficking between lysosomes and mitochondria drives a self-sustaining propagation of disrupted iron homeostasis and oxidative damage.
3.2. Intercellular: Synergistic Amplification via Contact-Dependent and Non-Contact Trigger Waves
Accumulating evidence from both in vitro and in vivo studies indicates that ferroptosis exhibits non-random, wave-like transmission characteristics in tissues [131,132]. Its mechanism includes two synergistic amplification pathways: contact-dependent lipid peroxidation transfer and non-contact trigger waves. The former occurs at cell–cell contact interfaces and relies on iron ions and oxidizable membrane lipids to extend the chain reaction of lipid peroxidation autocatalytically to adjacent plasma membranes, without the participation of intracellular components [133]. The latter originates from the damage of lipid peroxidation to the integrity of the cell plasma membrane, leading to cell swelling and the release of intermediate signaling molecules [132]. These molecules form self-sustaining trigger waves that can propagate long distances (>118 μm) without attenuation, inducing lipid peroxidation bursts and ferroptosis in neighboring cells [134].
It is worth noting that these two propagation modes exhibit selective dependence on the ferroptosis induction mechanism. Contact propagation has been demonstrated in the GPX4 inhibition model, but its universal chemical basis—lipid peroxidation being the core event in ferroptosis—suggests its potential broad applicability [18]. In contrast, trigger wave propagation has only been reported to occur under the condition of cysteine deficiency. Compared with the rapid lipid peroxidation initiation (within 1.5 h) in the GPX4 inhibition model, the slower progression and extensive ROS accumulation in erastin-induced ferroptosis provide the necessary time window and material basis for the production and release of intermediate mediators [135]. Although the exact molecular mediator remains elusive, key experimental evidence indicates that ROS is a diffusible signal: (1) conditioned medium containing factors secreted by ferroptotic cells can induce ferroptosis in recipient cells, indicating the presence of diffusible molecules; (2) cell death is greatly inhibited after pretreatment of this medium with ROS scavengers [134]. Consequently, the ferroptosis signal not only amplifies within cells but also propagates intercellularly via direct cell contact or diffusive molecules such as ROS, leading to widespread cell damage and population-level cell death.
4. Evolutionary Conservation in Development and Disease
To counteract oxidative damage, cells have evolved adaptive mechanisms to regulate intracellular lipid peroxidation. Lipid peroxidation is a radical-mediated cascade reaction in which molecular oxygen incorporates into lipids, generating lipid hydroperoxides via the peroxyl radicals’ intermediates. This process is ubiquitous in biological systems. Ferroptosis a form of iron-dependent regulated cell death driven by lipid peroxidation, is therefore hypothesized to be evolutionarily conserved across species (Figure 5).
Extensive studies in mouse models have firmly linked ferroptosis to the pathogenesis of multiple diseases, most prominently neurodegenerative disorders driven by pathogenic protein mutations. In models expressing SOD1^G93A^, TDP-43, or C9orf72, reduced GPX4 activity is tightly associated with ferroptotic lipid peroxidation, whereas genetic or pharmacological enhancement of GPX4 function significantly delays disease progression [136]. In Huntington’s disease mouse models driven by mutant huntingtin, pharmacological inhibition of ferroptosis with Liproxstatin-1 markedly reduces medium spiny neuron loss [137], while genetic deletion of ALOX5 suppresses ferroptosis, rescues disease phenotypes, and prolongs survival [33]. Synucleinopathies, which are neurodegenerative disorders characterized by the pathological aggregation of α-synuclein and include Lewy body disease, Parkinson’s disease, and multiple system atrophy, provide additional evidence linking ferroptosis to protein-misfolding neurodegeneration. Multiple mouse studies have demonstrated that aberrant α-synuclein accumulation can directly induce neuronal ferroptosis [138,139]. Consistently, in human iPSC-derived neurons, overexpression of SNCA, which encodes α-synuclein, significantly reduces cellular viability. Notably, ferroptosis inhibitors—including deferoxamine (an iron chelator), deuterated polyunsaturated fatty acids, and Ferrostatin-1—substantially attenuate α-synuclein oligomer-induced neuronal death and restore cellular function [140]. Further support for a causal role of ferroptosis in neurodegeneration comes from neuroferritinopathy, a genetic disorder caused by ferritin mutations. These mutations lead to profound iron dysregulation and robust activation of ferroptosis, which can be effectively mitigated by ferroptosis inhibitors [141]. Importantly, human genetic evidence underscores the central role of GPX4 in neuronal ferroptosis suppression. In Sedaghatian-type spondylometaphyseal dysplasia, the disease-causing GPX4^R152H^ mutation disrupts membrane targeting of GPX4. Despite retaining enzymatic activity, the mutant protein fails to localize properly to cellular membranes, rendering it incapable of suppressing lipid peroxidation. This defect results in uncontrolled neuronal ferroptosis and severe early-onset neurodegeneration [142], highlighting that GPX4-mediated ferroptosis protection critically depends on both enzymatic function and subcellular localization.
Beyond degenerative disorders, ferroptosis has emerged as a key determinant of therapeutic resistance in cancer, where reinforced antioxidant defenses and lipid metabolic remodeling enable tumor cells to escape ferroptosis under treatment pressure [128,129]. In parallel, ferroptosis plays a prominent role in inflammation and immune regulation, influencing immune cell survival, and inflammatory signaling [128,130]. Ferroptosis is also repeatedly implicated in murine ischemia–reperfusion injury, in which its inhibition confers substantial tissue protection [143]. Together, these disease models-many of which recapitulate core features of human pathology-support a conserved role for ferroptosis across neurodegeneration, cancer, inflammation, and acute tissue injury.
Recent studies have extended the relevance of ferroptosis beyond mammals to evolutionarily distant organisms. In Caenorhabditis elegans, aging is accompanied by pronounced ferroptotic features, and pharmacological inhibition of ferroptosis significantly extends lifespan [144]. Similarly, ferroptosis can be readily detected in C. elegans under specific stress conditions, including iron overload and dietary enrichment with PUFAs [145,146]. These observations indicate that the coupling between iron metabolism, PUFA oxidation, and ferroptotic cell death represents a deeply conserved biological vulnerability. The yolk of oviparous species is rich in PUFAs, which are essential for neural development but are highly susceptible to oxidative damage. To mitigate peroxidation-induced radical generation and lipid hydroperoxides formation, the oocyte system contains a complex array of antioxidants, including α-tocopherol [50,147], selenium [148], and antioxidant peptides, referring to small endogenous or dietary peptides capable of scavenging lipid radicals or supporting redox buffering [149]. The bioavailability of these nutrients is crucial during the development of oviparous organisms. For instance, selenium deficiency induces lipid peroxidation-related muscular dystrophy in chickens [150], while vitamin E deficiency in zebrafish embryos leads to dysregulation of thiols, amino acids, and related molecules [151], exacerbating lipid peroxidation and even leading to embryonic lethality [152]. Moreover, pharmacological inhibition of thiol-containing molecules can induce histological abnormalities and elevated malondialdehyde levels in zebrafish gonadal tissues [153]. Notably, studies have shown that broiler chickens under heat stress exhibit significant overproduction of ROS [154], which induces visceral oxidative damage [155] and activation of ferroptosis [156,157]. Emerging evidence also links ferroptosis to pathological manifestations of other species: novel duck reovirus (NDRV)-induced splenic lesions in ducklings and di(2-ethylhexyl) phthalate (DEHP)-mediated nephrotoxicity in quail (Coturnix japonica) [158,159]. These findings suggest that antioxidant defenses and ferroptosis pathways are involved in physiological and pathological processes in oviparous species.
Ferroptosis-like cell death processes have been widely reported in plants, such as Arabidopsis thaliana [160], rice plants [161,162], tobacco plants (N. benthamiana) [163], wheat roots (Triticum aestivum L.) [164], and faba bean roots (Vicia faba) [165] under heat stress and pathogen infection. Plant ferroptosis shares conserved molecular hallmarks with animal ferroptosis, including iron-dependent ROS accumulation, glutathione depletion, and lipid peroxidation, as well as morphological features such as cytoplasmic condensation, intact nuclei, and mitochondrial shrinkage [160]. In plants, ferroptosis-like cell death is generally considered maladaptive under abiotic stress conditions but may play a beneficial role in limiting pathogen spread during host–pathogen interactions, highlighting its context-dependent functional significance. While several reviews have outlined the landscape of ferroptosis in plants, a critical knowledge gap remains: the mechanisms governing ferroptosis-related signaling within and/or across species—particularly in host–pathogen interactions such as rice and M. oryzae—require systematic investigation to elucidate their regulatory networks and propagation dynamics [166,167,168].
Beyond plants, ferroptosis-like processes have also been observed in prokaryote Synechocystis sp. PCC 6803 [169] (which contains PUFA-rich thylakoid membranes despite prokaryotes generally lacking PUFAs in their membranes), unicellular algae Chlamydomonas reinhardtii [170], and fungal pathogens Magnaporthe oryzae [171]. Reviews by Conrad et al. [172] and Berndt et al. [173] described the roles and regulatory mechanisms of lipid peroxidation, ferroptosis, and antioxidant networks in yeast, bacteria, archaea, trypanosoma brucei, and drosophila melanogaster.
Although broader phylogenetic validation is warranted, current evidence underscores the evolutionary conservation of ferroptosis, which is involved in critical biological processes such as developmental regulation, stress adaptation, disease pathogenesis, and drug responses. These conserved pathways highlight ferroptosis as a universal cell death modality with profound implications for both fundamental biology and translational research.
Evidence suggests that while the ferroptosis program is explicitly defined in mammals, the core machinery of lipid peroxidation and associated antioxidant pathways display significant evolutionary conservation across plants, oviparous animals, and even microorganisms, indicating its ancient and fundamental role in biology.
5. Discussion
Ferroptosis, a regulated form of cell death, has emerged as an active field of research characterized by several distinctive features. Mechanistically, the dynamic interplay between lipid peroxidation and the antioxidant defense network confers plasticity to ferroptosis. lipid peroxidation can be initiated through different pathways and is influenced by substrate availability, whereas the antioxidant system adapts dynamically through a core-compensatory dual hub, expansion of the antioxidant molecule pool, and transcriptional reprogramming. The plasticity enables cells to maintain redox homeostasis under stress or nutrient imbalance, but also constitutes a foundation for drug resistance. Combination therapeutic strategies that simultaneously target multiple key nodes hold promise for overcoming current treatment limitations. Intracellular and extracellular Lipid peroxidation and ferroptosis-related signals/molecules form a spatially coordinated network, amplifying cell death effects exponentially across cell populations. This propagation system explains the widespread propagation of tissue damage and rapid pathological progression, highlighting its promise for novel therapeutic interventions in diseases like IRIs and neurodegenerative disorders. From an evolutionary perspective, ferroptosis(-like) and antioxidant processes are widely conserved across mammals, oviparous species, plants, and microorganisms, contributing to developmental homeostasis and host–pathogen interactions. These insights may inspire novel strategies for managing infectious diseases or modulating anti-tumor immunity.
Growing evidence suggests that in specific histological contexts or pathological states characterized by metabolic hyperactivity, lipid enrichment, or iron overload, ferroptosis may play a more prominent or earlier role compared to other regulated cell death (RCD) modalities, such as apoptosis, necroptosis, and pyroptosis [174]. In acute organ failure and IRI, ferroptosis often emerges as a prominent contributor [175,176,177]. Particularly in the kidneys and heart, the reperfusion-induced oxidative burst directly triggers membrane lipid peroxidation, leading to cell death before the activation of apoptotic pathways [178,179]. Studies in models of acute kidney injury, myocardial infarction, and stroke have demonstrated that inhibiting ferroptosis significantly attenuates tissue damage and improves functional recovery [180,181,182]. This is particularly salient in organ transplantation, where ferroptosis serves as a major lethal event post-reperfusion, positioning it as a promising therapeutic target [176]. Similarly, the brain is inherently susceptible to ferroptosis due to its high concentration of PUFAs and substantial accumulation of iron [183]. In neurodegenerative disorders like Parkinson’s and Alzheimer’s, aberrant protein aggregation, dysregulated iron metabolism, and chronic oxidative stress provide a sustained impetus for ferroptotic signaling [184]. Furthermore, mesenchymal-state or mutant (e.g., KRAS, TP53) cancer cells exhibit hypersensitivity to ferroptosis [11,185,186]. Metabolic reprogramming during epithelial–mesenchymal transition allows drug-resistant cells to evade apoptosis while simultaneously creating a ferroptosis-sensitive vulnerability, offering a strategy for treating recalcitrant malignancies like glioma [187,188,189].
Nevertheless, caution is warranted regarding the conclusion that ferroptosis is “dominant.” Ferroptosis is not an independent process but is deeply embedded within a complex regulatory network of RCD. Central to this network are ROS, which act as both the primary executioners of ferroptosis and the essential signaling conduits between diverse RCD pathways [190,191]. The accumulation of lipid-derived ROS triggers ferroptosis, whereas ROS compartmentalization and flux levels dictate the initiation of apoptosis, autophagy, necroptosis, or pyroptosis [192,193,194,195]. Concurrently, several key molecules function as “hubs” connecting different death pathways. For example, TP53, a classical pro-apoptotic gene, promotes apoptosis by activating p53 upregulated modulator of apoptosis (PUMA), Phorbol-12-myristate-13-acetate-induced protein 1 (PMAIP1), and other effectors [196,197]. It also acts as a multifaceted fate determinant by suppressing SLC7A11 to sensitize cells to ferroptosis, upregulating necrosis-related factor (NRF) to facilitate necroptosis, and activating expressions of pyroptosis regulators such as the NACHT, LRR and PYD domains-containing protein 3 (NLRP3), caspase 1, and gasdermin E (GSDME) [198]. Therefore, the interactions between ferroptosis and other cell death modalities are diverse and context-dependent. Specifically, ferroptosis and apoptosis exhibit a complex interplay of competition and synergism. On one hand, fatty acid oxidation activates STAT3 through CoA acetylation and upregulates the expression of ACSL4, a key ferroptosis-relate enzyme, enabling tumor cells to evade chemotherapy-induced apoptosis [199]. On the other hand, ferroptosis-induced mitochondrial dysfunction and ROS accumulation are known to trigger mitochondrial outer membrane permeabilization (MOMP), a definitive hallmark of the intrinsic apoptotic pathway [200]. Consequently, clinical strategies now increasingly aim to co-induce these two pathways to overcome therapeutic resistance [201]. Furthermore, ferroptosis and necroptosis share ROS storms and often occur synergistically. Since ROS simultaneously drives lipid peroxidation and activates receptor-interacting protein kinase 1 (RIPK1), these pathways serve as parallel and complementary clearance mechanisms in pathologies such as acute kidney injury [202]. There is a stronger link between ferroptosis and autophagy (a lysosomal-dependent process). As noted, selective autophagy, particularly NCOA4-mediated ferritinophagy, mobilizes the labile iron pool via ferritin degradation, thereby potently driving ferroptotic execution. Similarly, lipophagy provides the essential lipid substrates for peroxidation, positioning autophagic flux as a critical rheostat for ferroptotic sensitivity [203]. Moreover, ferroptosis and pyroptosis establish a “metabolic-to-immunogenic death” cascade. Damage-associated molecular patterns (DAMPs) released by ferroptotic cells activate the NACHT, NLRP3 inflammasome, triggering pyroptosis and the subsequent release of pro-inflammatory cytokines, which amplifies tissue damage or anti-tumor immunity [204,205]. Additionally, ferroptosis intersects with emerging cuproptosis through mitochondrial metabolic stress and metal toxicity [206], and with paraptosis via disrupted redox and ionic homeostasis—both converging on mitochondrial dysfunction as a shared execution node [207]. In conclusion, ferroptosis integrates with other RCD programs to form an interconnected decision-making network, orchestrated by shared ROS signaling, universal molecular hubs, and dynamic transition mechanisms. Given that most diseases involve a complex interplay of multiple RCD subroutines, the perceived “dominance” of ferroptosis may be confined to specific spatiotemporal windows or cellular subpopulations. Moreover, current evidence relies heavily on the rescue effects of inhibitors like Ferrostatin-1, which may exert non-specific effects on other ROS-dependent pathways, necessitating careful interpretation of the “dominance”. Ultimately, while characterizing a single modality as dominant may oversimplify the dynamic death network, targeting ferroptosis remains an indisputably promising therapeutic approach.
However, many mechanistic details remain incompletely understood. Emerging evidence suggests that lipid peroxides initially accumulate in organelles, such as the endoplasmic reticulum, before propagating to the plasma membrane [18,208], where they activate mechanosensitive ion channels including Piezo1 and transient receptor potential (TRP) channels [209]. This channel activation disrupts ion homeostasis, ultimately leading to membrane rupture. However, how lipid peroxidation precisely modulates membrane biophysical properties, and why interventions such as polyethylene glycol only delay but not prevent rupture [210], indicate the involvement of multiple contributing factors. Future studies could employ super-resolution imaging to visualize the dynamics of membrane nanopore formation and explore specific inhibitors targeting mechanosensitive channels to postpone membrane disintegration.
Furthermore, the compartmentalized regulation of lipid metabolism—such as the synthesis and trafficking of phosphatidylethanolamine—and the precise subcellular localization of key enzymes like ACSL4 remain unclear [211]. Interorganellar communication via membrane contact sites may facilitate the spread of lipid peroxidation, although the underlying mechanisms are not yet defined. Mitochondria and lipid droplets appear to play context-dependent roles in ferroptosis, with their contributions varying across cell types and microenvironments. Establishing conditional models that account for the functional heterogeneity of these organelles may help resolve existing controversies.
Iron metabolism represents another critical layer of regulation. The contributions of various iron sources—including transferrin-bound iron, iron-sulfur clusters, and heme—as well as iron release mechanisms such as ferritinophagy and quinone-mediated pathways [212], are not fully elucidated, particularly in mammalian systems [213]. In physiological and pathological contexts, ferroptosis is involved in development, aging, immune surveillance, and infection. How pathogens exploit ferroptosis to promote their survival, and how hosts counteract this process, remain important areas of investigation. Developing combination therapies involving ferroptosis inhibitors together with antibiotics or immunotherapies requires careful evaluation of potential benefits and risks. Notably, ferroptosis exerts dual effects in immune regulation: it can enhance antitumor immunity but may also suppress immune tolerance [214]. Its precise modulation poses a challenge for cancer therapy. Current research often focuses on how immune cells such as T cells and macrophages regulate ferroptosis sensitivity via cytokines like IFNγ [215,216,217,218]. Therefore, establishing reliable in vivo models to dissect the complex immune microenvironment is essential. Moreover, given that ferroptosis involves intercellular propagation and feedback mechanisms, organoid models may serve as an ideal system for further exploration.
Although ferroptosis initiation is of fundamental importance, accumulating evidence suggests that its onset is governed by multiple, context-dependent mechanisms, potentially involving coordinated redox reactions, iron handling, lipid metabolic remodeling, and upstream signaling pathways such as kinase cascades [173]. Because these early triggering events remain incompletely defined and are actively debated, they are not discussed in detail here. Instead, this Review focuses on the downstream amplification, execution, and propagation of ferroptosis damage, while highlighting ferroptosis initiation as a critical and open area for future investigation.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Stockwell B.R. Friedmann Angeli J.P. Bayir H. Bush A.I. Conrad M. Dixon S.J. Fulda S. Gascón S. Hatzios S.K. Kagan V.E. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease Cell 201717127328510.1016/j.cell.2017.09.02128985560 PMC 5685180 · doi ↗ · pubmed ↗
- 2Dixon S.J. Lemberg K.M. Lamprecht M.R. Skouta R. Zaitsev E.M. Gleason C.E. Patel D.N. Bauer A.J. Cantley A.M. Yang W.S. Ferroptosis: An iron-dependent form of nonapoptotic cell death Cell 20121491060107210.1016/j.cell.2012.03.04222632970 PMC 3367386 · doi ↗ · pubmed ↗
- 3Dixon S.J. Olzmann J.A. The cell biology of ferroptosis Nat. Rev. Mol. Cell Biol.20242542444210.1038/s 41580-024-00703-538366038 PMC 12187608 · doi ↗ · pubmed ↗
- 4Riegman M. Bradbury M.S. Overholtzer M. Population Dynamics in Cell Death: Mechanisms of Propagation Trends Cancer 2019555856810.1016/j.trecan.2019.07.00831474361 PMC 7310667 · doi ↗ · pubmed ↗
- 5Lin Y. Xu W. Hou Y. Wang S. Zhang H. Ran M. Huang Y. Wang Y. Yang G. The multifaceted role of ferroptosis in kidney diseases Chem. Biol. Interact.202236511010710.1016/j.cbi.2022.11010735985518 · doi ↗ · pubmed ↗
- 6Zhou Y. Lin W. Rao T. Zheng J. Zhang T. Zhang M. Lin Z. Ferroptosis and Its Potential Role in the Nervous System Diseases J. Inflamm. Res.2022151555157410.2147/JIR.S 35179935264867 PMC 8901225 · doi ↗ · pubmed ↗
- 7Shrivastav D. Mishra J. Sharma V.K. Singh S. Khan M.I. Alsanie S.A. Ashfaq F. Beg M.M.A. Biochemical and Physiological Response During Oxidative Stress: A Cross-Species Perspective Rejuvenation Res.202510.1177/1549168425138671241069197 · doi ↗ · pubmed ↗
- 8Huang Q. Ru Y. Luo Y. Luo X. Liu D. Ma Y. Zhou X. Linghu M. Xu W. Gao F. Identification of a targeted ACSL 4 inhibitor to treat ferroptosis-related diseases Sci. Adv.202410 eadk 120010.1126/sciadv.adk 120038552012 PMC 10980261 · doi ↗ · pubmed ↗