Characteristics and Phylogenetic Considerations of the Newly Sequenced Mitochondrial Genome of Teratoscincus scincus (Gekkota: Sphaerodactylidae)
Zhiqiang Ge, Zhengyu Zhang, Zelu Mu, Linqiang Zhong

TL;DR
This paper reports the sequencing of the mitochondrial genome of Teratoscincus scincus and uses it to clarify its evolutionary relationships within the Sphaerodactylidae family.
Contribution
The study provides the first complete mitochondrial genome of T. scincus and confirms its phylogenetic placement within the genus Teratoscincus.
Findings
The mitochondrial genome of T. scincus is 16,943 bp long and has a high A + T content of 56.3% in protein-coding genes.
Phylogenetic analysis shows T. scincus forms a monophyletic clade with T. keyserlingii, supporting its classification in the genus Teratoscincus.
The study enriches the mitochondrial genomic database for Sphaerodactylidae and supports future research on adaptive evolution and conservation.
Abstract
This study sequenced and analyzed the complete mitochondrial genome of Teratoscincus scincus using the Illumina NovaSeq Xplus platform. The circular mitogenome is 16,943 bp in length, comprising 13 protein-coding genes (PCGs), 22 tRNA genes, 2 rRNA genes, and 1 control region. It exhibits a distinct AT preference, with the highest A + T content (56.3%) observed in the PCGs. Phylogenetic analysis based on 13 PCGs from 10 Sphaerodactylidae species confirmed that T. scincus belongs to the genus Teratoscincus, forming a monophyletic clade with Teratoscincus keyserlingii. This work enriches the mitochondrial genomic database for Sphaerodactylidae and lays a foundation for research on the adaptive evolution and conservation of T. scincus. Sphaerodactylidae play a crucial role in ecosystems, possessing significant ecological, scientific, and conservation value. They contribute to pest control…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
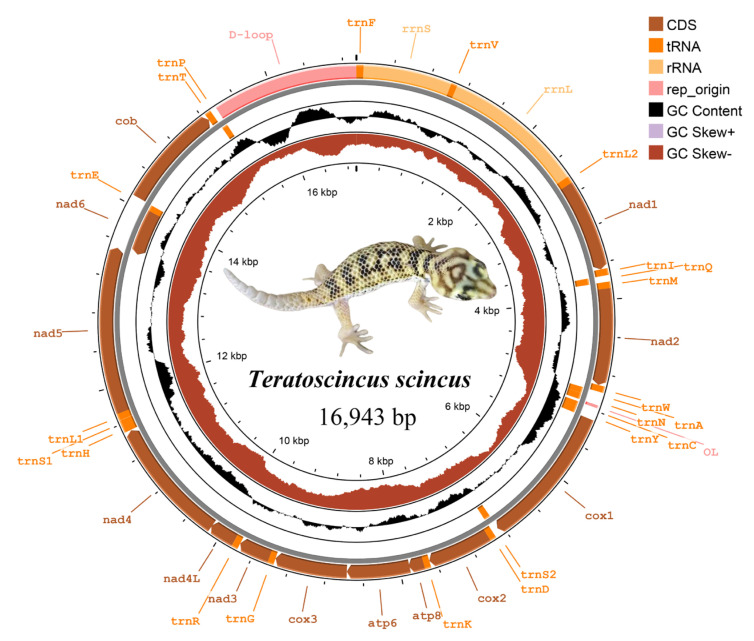 Figure 1
Figure 1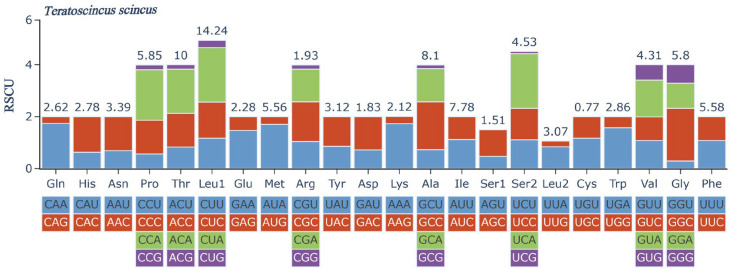 Figure 2
Figure 2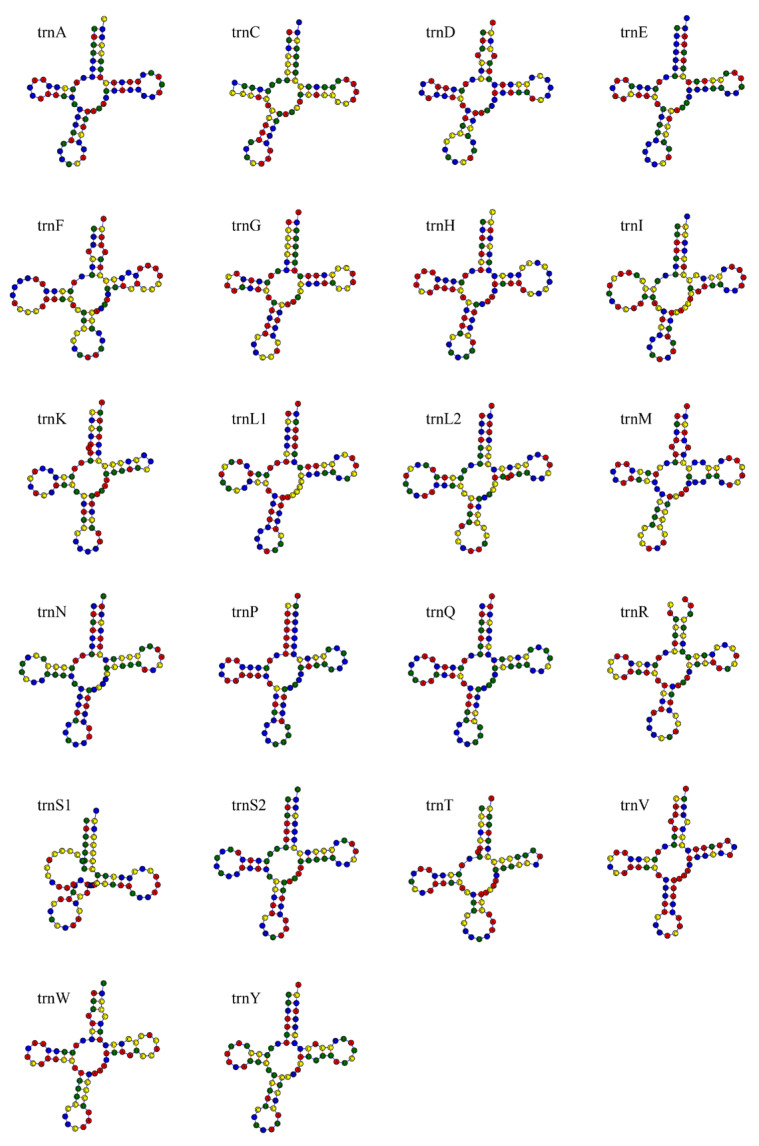 Figure 3
Figure 3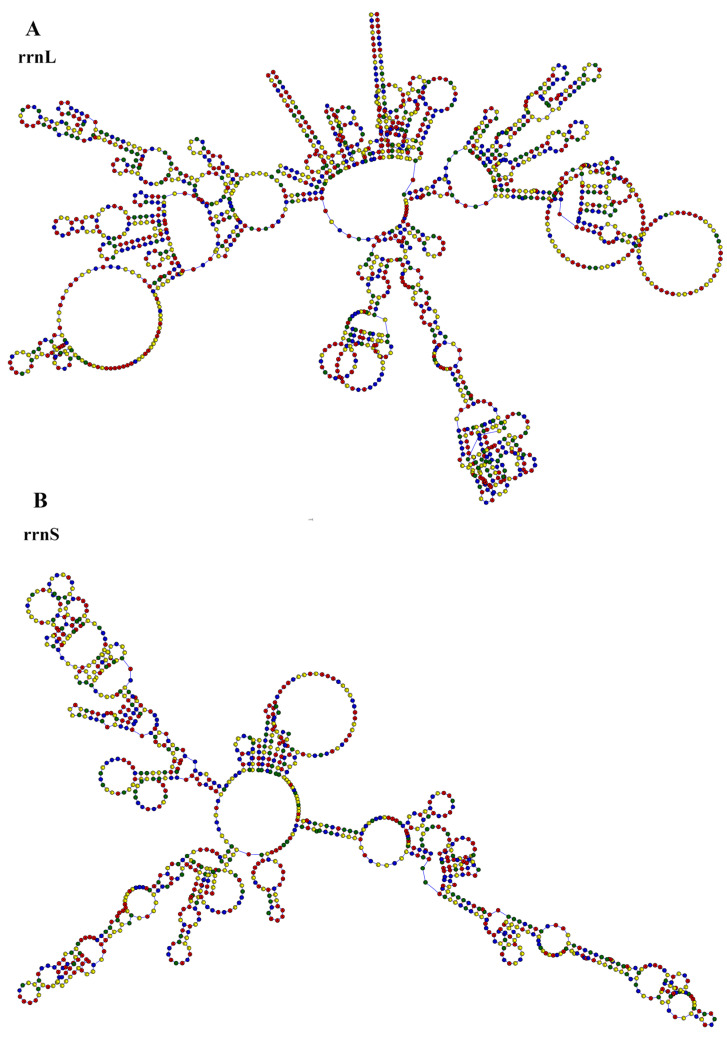 Figure 4
Figure 4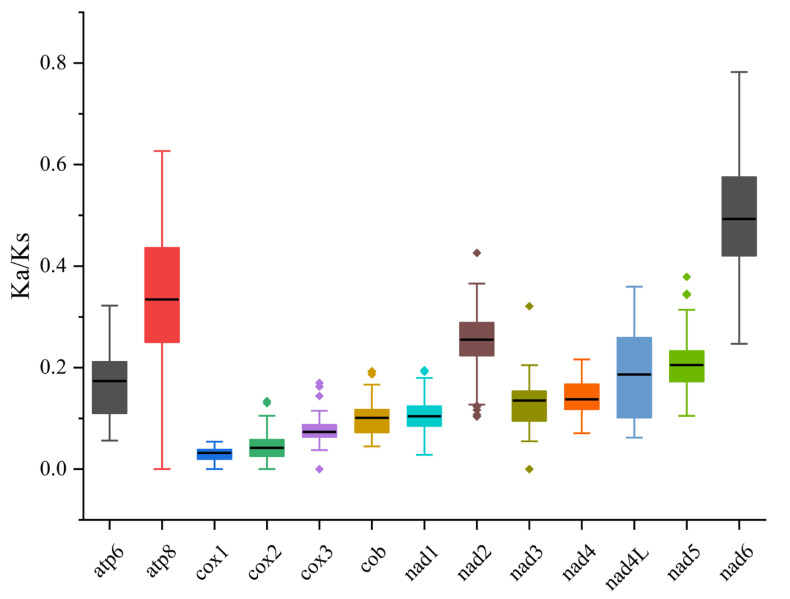 Figure 5
Figure 5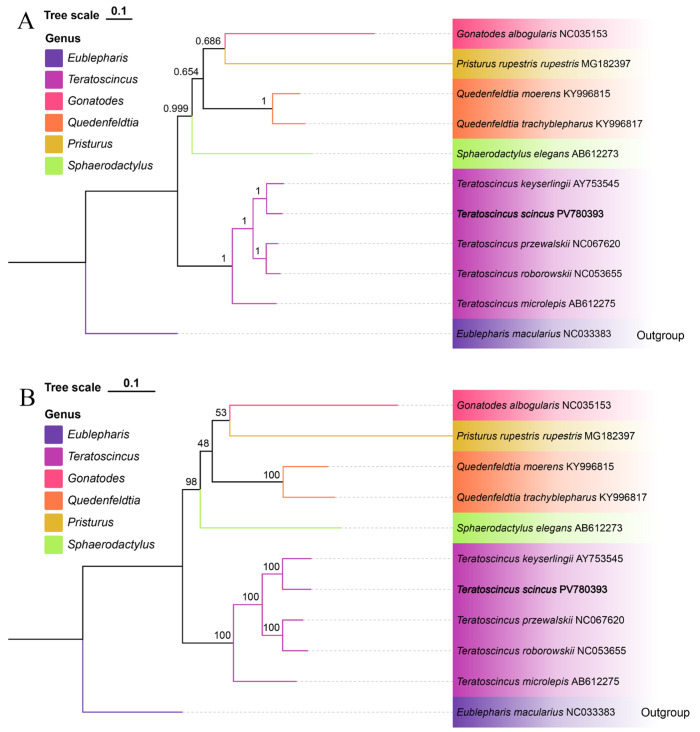 Figure 6
Figure 6- —Natural Science Foundation of Xinjiang Uygur Autonomous Region
- —Third Xinjiang Scientific Expedition Program
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsGenomics and Phylogenetic Studies · Amphibian and Reptile Biology · Lepidoptera: Biology and Taxonomy
1. Introduction
The Sphaerodactylidae, belonging to the Squamata, are a group of small to micro-sized geckos. Currently, this family is known to include 12 genera and over 200 species [1]. This group is characterized by slender body shapes, specialized ecological niches, and a wide distribution in tropical and subtropical regions, mainly concentrated in the Americas and the Caribbean; however, only a few lineages (e.g., Teratoscincus) have dispersed to the arid regions of Central Asia [2,3].
Teratoscincus is a representative genus of the Sphaerodactylidae in the arid and semi-arid regions of Central Asia and the Middle East [4]. This genus was initially classified under the Gekkonidae but was later reclassified into the Sphaerodactylidae based on molecular, morphological, and anatomical evidence [5,6]. Currently, nine species are recognized in this genus [4], three of which are distributed in China: Teratoscincus roborowskii, Teratoscincus przewalskii, and Teratoscincus scincus [3]. The distribution range of T. scincus is highly fragmented, and habitat degradation is increasingly severe; in border areas, in particular, the fragmentation of conservation policies further increases its survival risks [7].
The mitochondrial genome (mtDNA) is an essential genetic material in cells [8], exhibiting unique genetic characteristics such as maternal inheritance, a high mutation rate, and gene rearrangement [9]. It has become an ideal model for studying population genetic structure, molecular evolutionary processes, and interspecific genetic relationships in animals [10]. In most metazoans, the mitochondrial genome is a small closed circular double-stranded DNA molecule, usually containing 13 PCGs, 22 tRNA genes, and 2 rRNA genes [11,12]. In the early stage of analyzing the composition and structural characteristics of mitochondrial genomes, research mainly relied on Sanger sequencing technology [13]; in recent years, with the development of bioinformatics technology, the assembly, annotation, and analysis of mitochondrial genome sequences have been significantly improved in terms of accuracy and efficiency [14]. Molecular phylogenetic analysis based on mitochondrial genes effectively compensates for the limitations of traditional morphological classification in the identification of closely related species. Compared with phylogenetic analysis based on single genes, the multi-gene combined analysis strategy can provide more abundant genetic information and has thus become the mainstream method in phylogenetic relationship reconstruction [15].
However, there remains a significant gap in our understanding of the mitochondrial genomes of Sphaerodactylidae, particularly the arid-adapted genus Teratoscincus. Although T. scincus is one of the species distributed in China, its complete mitochondrial genome has not yet been sequenced, which has hindered in-depth phylogenetic and adaptive evolution studies on both the genus and the entire family. To fill this data gap, in this study, we sequenced, annotated, and analyzed the structural characteristics of the mitochondrial genome of T. scincus and constructed a phylogenetic tree. This work not only enriches the mitochondrial genome database of Sphaerodactylidae but also provides important references for the taxonomic and evolutionary research of species in this family. Furthermore, it offers a theoretical basis and data support for species diversity conservation and germplasm resource evaluation.
2. Materials and Methods
2.1. Sample Collection
A single T. scincus sample was collected from Huocheng County, Xinjiang Uygur Autonomous Region, China (Table 1). This sample was a roadkill carcass obtained during animal surveys. For preliminary morphological identification, we followed the key taxonomic characters of the genus Teratoscincus and species-specific diagnostic traits: cycloid scales of the back are strongly imbricate, extending onto the posterior part of the head, with 28–34 scales around the midbody [16]. After confirming the sample’s taxonomic identity based on these morphological characters, tail tissue was collected, stored in a 1.5 mL centrifuge tube, and preserved in a −80 °C ultra-low temperature freezer for subsequent use.
2.2. DNA Extraction
Genomic DNA was extracted from muscle tissue using the Blood/Cell/Tissue Genomic DNA Extraction Kit (TIANamp Genomic DNA Kit) purchased from TIANGEN BIOTECH (Beijing, China) Co., Ltd, following the manufacturer’s instructions and stored in a −20 °C freezer. To verify the accuracy of morphological identification, the mitochondrial cox1 gene of T. scincus was amplified using the primers LOC1490 (5′-GGTCACAACAAATCATAAAGATATTGG-3′) and HOC2198 (5′-TAAACTTCAGGGTGACCAAAAAATCA-3′) [17]. The PCR system (25 μL) consisted of 12.5 μL of PreMix Taq enzyme, 0.5 μL of each primer (forward and reverse), 5 μL of the DNA template, and 6.5 μL of double-distilled water (ddH2O) [18]. The specific PCR conditions for the cox1 gene are shown in Table 2. The PCR product was sent to Sangon Biotech (Shanghai, China) Co., Ltd. for Sanger sequencing. The sequencing results were subjected to BLAST v2.17.0 comparison through NCBI, and the comparison results confirmed the species as T. scincus.
2.3. Mitochondrial Genome Sequencing and Data Filtering
The muscle tissue sample of T. scincus was sent to Beijing Berry Genomics Co., Ltd. (Beijing, China). A genomic DNA library with an insert size of approximately 350 bp was constructed using the Illumina NovaSeq Xplus platform (Illumina, San Diego, CA, USA), and paired-end 150 bp (PE150) sequencing was performed to obtain approximately 6 GB of raw data (in FASTQ format). Subsequently, fastp v0.23.4 [19] with parameters -f 5 -t 5 -F 5 -T 5 -5 -3 -W 4 -M 20 -c -q 15 -u 40 -l 35 was used for quality control and filtering of raw reads to remove reads containing adapter sequences, high repetition rates, high N content, and low quality, thereby obtaining high-quality data for subsequent assembly.
2.4. Mitochondrial Genome Assembly, Annotation, and Validation
To ensure the accuracy of results, multiple strategies were adopted for the assembly and validation of the mitochondrial genome in this study. First, MitoZ v3.6 [20] was used for the integrated processing of raw sequencing data, including quality control, de novo assembly, and preliminary annotation, with the following key parameters: --clade Chordata, --genetic_code 2, --fastq_read_length 151, --data_size_for_mt_assembly 0, --assembler megahit (configured with k-mer sizes of 71 and 99), --memory 50, and --requiring_taxa Chordata. For cross-validation, high-quality reads filtered by fastp were imported into Geneious Prime v2025.1.2, and the “Map to Reference” function was used for secondary assembly. Then, the two assembly results were aligned and manually checked in MEGA v11 [21] to finally obtain a consistent high-quality mitochondrial genome sequence. Meanwhile, MITOS2 v2.1.9 [22] and tRNAscan-SE v2.0 [23] were used for gene prediction and tRNA secondary structure prediction of the T. scincus mitochondrial genome, respectively. The annotation results from MitoZ v3.6 and MITOS2 v2.1.9 were manually verified in Geneious Prime for final confirmation. Based on the verified genome sequence, a circular map of the mitochondrial genome was generated using the Proksee platform [24]. The final assembled sequence and annotation information have been submitted to the NCBI GenBank database with the accession number PV780393.
2.5. Sequence Analysis
To analyze the sequence characteristics of the mitochondrial genome, first, MEGA v11 [21] software was used to calculate the base content of the entire genome, and the nucleotide skewness was calculated according to the following skewness formulas: “AT-skew = (A − T)/(A + T)” and “GC-skew = (G − C)/(G + C)” [25]. Second, DNAsp v6.0 software was used to conduct an in-depth analysis of the evolutionary characteristics of the genome using the Ka/Ks ratio (non-synonymous/synonymous substitution rate ratio) as an indicator. In addition, PhyloSuite v1.2.3 [26,27] software was used to calculate the relative synonymous codon usage (RSCU) of PCGs, and the results of codon usage analysis were visualized.
2.6. Phylogenetic Analysis
Based on Bayesian inference and maximum likelihood methods, the effective mitochondrial genomes of 10 Sphaerodactylidae species and the mitochondrial genome of Eublepharis macularius (NC033383) as the outgroup were selected to conduct phylogenetic analysis of their 13 PCGs (Table 3). All analyses were completed on the PhyloSuite v1.2.3 [26,27] platform. The specific workflow was as follows: First, the corresponding GenBank format sequences were downloaded from the NCBI database and imported into PhyloSuite v1.2.3 for gene screening and annotation. MAFFT v7.526 was used for multiple sequence alignment [28]; then, MACSE v2 [29] was used to optimize the alignment results, followed by trimming with trimAl v2 [30] to obtain optimized aligned sequences. FASconCAT-G v1.04 [31] was used to concatenate the sequences. IQ-TREE v2.2.0 [32] was used to construct the ML tree. Under the Akaike information criterion, ModelFinder v2.2.0 was used to determine the optimal partitioning scheme (edge-linked) and substitution model, and 1000 bootstrap replicates were used to evaluate the confidence values of each node in the phylogenetic tree [33]. MrBayes v3.2.7 [34] with 2 independent runs was used to construct the BI tree, with each run having 4 Markov chain Monte Carlo (MCMC) chains of 5,000,000 generations, sampled every 1000 generations. The first 25% of the runs were discarded to estimate the posterior probabilities of branch confidence. ModelFinder v2.2.0 and the corrected Bayesian information criterion were used to determine the optimal partitioning scheme (edge-linked) and model. Finally, the online software ChiPlot v1 was used to visualize and refine the phylogenetic tree [35].
3. Results
3.1. Genome Organization and Base Composition
The complete mitochondrial genome of T. scincus obtained via sequencing is a typical double-stranded circular DNA molecule with a size of 16,943 bp (Figure 1). The mitochondrial genome of T. scincus sequenced in this study contains 13 PCGs, 22 transfer RNA (tRNA) genes, 2 ribosomal RNA (rRNA) genes, and 1 non-coding sequence (D-loop region). Eight tRNA genes (trnQ, trnA, trnN, trnC, trnY, trnS2, trnE, trnP) and nad6 are located on the L-strand, while the remaining genes are on the H-strand (Table 4). There are eight gene overlaps and six gene intervals in the mitochondrial genome (Table 4).
The gene overlaps are located between trnI and trnQ (−1 bp), trnQ and trnM (−1 bp), trnW and trnA (−1 bp), OL and trnC (−2 bp), cox1 and trnS2 (−9 bp), atp8 and atp6 (−10 bp), nad4L and nad4 (−7 bp), and nad5 and nad6 (−8 bp), with the longest sequence being 10 bp and the shortest 1 bp. The gene intervals are located between trnA and trnN (1 bp), trnY and cox1 (1 bp), trnR and nad4L (1 bp), nad4 and trnH (5 bp), trnS1 and trnL1 (2 bp), and trnE and cob (3 bp), with the longest sequence being 5 bp and the shortest 1 bp.
In the complete mitochondrial genome of T. scincus, the contents of bases A, T, G, and C are 30.2%, 25.6%, 14.5%, and 29.7%, respectively, and the content of A + T (55.8%) is significantly higher than that of C + G (44.2%). Nucleotide composition analysis showed that T. scincus has a significant preference for AT nucleotides, i.e., there is AT bias in the mitochondrial genome. The AT-skew (0.082) is greater than the GC-skew (−0.345), and this pattern is also evident in the PCGs (AT-skew: 0.055 > GC-skew: −0.406), tRNAs (AT-skew: 0.110 > GC-skew: −0.204), and rRNAs (AT-skew: 0.230 > GC-skew: −0.221). The A + T content of PCGs in the mitochondrial genome is the highest (56.3%) (Table 5).
3.2. Protein-Coding Genes and Codon Usage Patterns
The total length of the 13 PCGs in the T. scincus mitochondrial genome is 11,367 bp (Table 5). Except for nad6, which is encoded on the L-strand, the others are encoded on the H-strand (Table 4). Among the PCGs of the mitochondrial genome, atp8 is the shortest (165 bp) and nad5 is the longest (1803 bp) (Table 4). In addition, nad1 starts with the ATC codon, nad2 with ATA, nad3 with ATT, nad5, cox1, and atp8 with GTG, and the remaining start codons are ATG (Table 4).
Five types of stop codons were identified in the PCGs of T. scincus, including incomplete codons T and TA, and complete codons TAA, TAG, and AGG (Table 4). nad2 and cox3 use T as the stop codon, cox1 uses AGG, atp6, cob, and nad3 use TA, nad1, nad4L, nad4, nad6, and atp8 use TAA, and nad5 uses TAG (Table 4). Among them, the TAA stop codon has the highest frequency of occurrence, while TAG and AGG have the lowest (Table 4). Through RSCU analysis, we studied the codon usage pattern of the T. scincus mitochondrial genome (Figure 2). The RSCU value of codon UCA is the highest (2.11), and that of UCG is the lowest (0.08) (Table 6).
3.3. Transfer RNAs, Ribosomal RNAs, and Control Region
A total of 22 tRNA genes were identified in the T. scincus mitochondrial genome, with each gene ranging in length from 66 to 75 bp, totaling 1532 bp. Fourteen tRNA genes are located on the H-strand, and eight on the L-strand. Among them, 21 tRNA genes can fold into the typical cloverleaf secondary structure, but the DHU arm of trnS1 is missing and fails to form a typical stem-loop structure (Figure 3).
The T. scincus mitochondrial genome contains a typical ribosomal RNA (rRNA) gene cluster, consisting of rrnL (1524 bp) (Figure 4A) and rrnS (949 bp) (Figure 4B), with a total length of 2473 bp. These two rRNA genes are located between the trnF (75 bp) and trnL2 (75 bp) genes, separated by the trnV (66 bp) gene. OL is located between the trnN and trnC genes, with a length of 31 bp (Table 4); the D-loop region is located between the trnP and trnF genes, with a length of 1566 bp (Table 4).
3.4. Synonymous and Non-Synonymous Substitution Rates
To study the evolutionary patterns and rates of mitochondrial PCGs in Sphaerodactylidae species, this study calculated the Ka/Ks ratios of homologous gene pairs of mitochondrial PCGs from 10 Sphaerodactylidae species and analyzed them in combination with the Ka/Ks values (Figure 5). The results showed that the average Ka/Ks ratios of the 13 PCGs in descending order are nad6, atp8, nad2, nad5, nad4L, atp6, nad4, nad3, nad1, cob, cox3, cox2 and cox1. The Ka/Ks ratio for all 13 PCGs was found to be below 1.
3.5. Phylogenetic Relationships
Using E. macularius (NC033383) as the outgroup, combined with the gene sequence of T. scincus (PV780393) sequenced in this study, phylogenetic analysis was conducted on the 13 PCG sequences of 9 Sphaerodactylidae species published in GenBank (Table 3). The phylogenetic tree showed that all Teratoscincus species clustered into a monophyletic group with high support, confirming the taxonomic status of this genus within Sphaerodactylidae. Within this genus, T. scincus and Teratoscincus keyserlingii clustered into one clade with high support (BI/ML > 99%), while T. przewalskii and T. roborowskii formed another monophyletic group, and Teratoscincus microlepis was the sister group of the above four species.
4. Discussion
In this study, we described and analyzed the complete mitochondrial genome of T. scincus, and examined its structural features, evolutionary patterns, and phylogenetic position within Sphaerodactylidae. It has a typical vertebrate structure with a length of 16,943 bp (13 PCGs, 22 tRNAs, 2 rRNAs, 1 D-loop) [7,36]. This work fills an existing gap in mitochondrial genomic data for this species and provides a useful reference for comparative mitogenomic and phylogenetic studies within the family.
Eight gene overlaps were observed in the T. scincus mitochondrial genome, among which the overlap region between atp8 and atp6 is the largest. This may facilitate the co-transcription and coordinated expression of these two functionally related genes, reflecting the economy of the mitochondrial genome [11,37]. Gene overlap not only saves bases and efficiently uses DNA but also may participate in gene regulation, allowing limited DNA to carry more genetic information, which reflects the rational use of genetic material by organisms [38]. This pattern may be consistent with adaptations to energy constraints in arid environments where resources are scarce, although direct functional tests are required.
The T. scincus mitochondrial genome shows a significant AT bias (A + T = 55.8%), with the highest AT content (56.3%) in the PCGs region (Table 5). This bias originates from the asymmetric replication of mitochondria [39], which can reduce the stability of DNA double strands and accelerate mutation accumulation, providing a genetic basis for the rapid generation of adaptive variations in arid environments [40,41].
The start and stop codon usage patterns of PCGs in T. scincus have certain particularities, and these variations may affect their transcription and translation efficiency, thereby playing a role in the evolutionary process [42,43,44]. Studies have found that in reptiles, ATG is the most common start codon, and some genes use incomplete stop codons (e.g., T and TA), which may be related to their living environment and physiological needs. Incomplete stop codons (e.g., T and TA) may complete the termination function through polyadenylation. Meanwhile, codon usage patterns can be used to distinguish different groups, and the frequent use of codons A and T with an obvious AT skew may be related to the compositional asymmetry of the mitochondrial genome, which also provides a genetic basis for T. scincus to rapidly generate adaptive variations in response to changes in arid environments [45].
The transcription of T. scincus mitochondrial genes is mainly based on the H-strand, while some genes are transcribed by the L-strand. This double-strand transcription mode ensures the efficient expression of mitochondrial genes [46]. tRNA genes are asymmetrically distributed on the two strands, with 14 tRNA genes located on the heavy strand (H-strand) and 8 on the light strand (L-strand). This distribution pattern is consistent with the typical characteristics of vertebrate mitochondrial genomes, ensuring the efficient transcription and translation of mitochondrial genes [47]. Twenty-one tRNA genes can fold into the typical cloverleaf secondary structure, which is crucial for maintaining the normal function of the mitochondrial protein synthesis system [47]. The genome shows a typical gene arrangement pattern of trnF/rrnS/trnV/rrnL/trnL2, which is widely present in vertebrate mitochondrial genomes [11]. The structural characteristics of rRNAs in T. scincus (the secondary structures of rrnS and rrnL are composed of multiple stem-loops, and rrnL is more complex than rrnS) may reflect adaptive changes during its evolution [48]. The non-coding regions include OL and D-loop. Among them, OL mainly functions to initiate the replication of the L-strand, while the D-loop is used to regulate transcription and initiate the replication of the H-strand. The two cooperate to maintain the stable replication and expression of the mitochondrial genome [49].
The adaptive characteristics of Sphaerodactylidae in morphology, behavior, and molecular levels are closely related to environmental pressures [3]. Nad6 is a core subunit of mitochondrial respiratory chain complex I (NADH dehydrogenase), whose core function is to participate in electron transfer and energy generation in cellular aerobic respiration [50]. The results suggest that nad6 is a fast-evolving gene, potentially driven by adaptive selection or relaxed constraints [51]. In extreme environments with scarce resources and high environmental pressure, species need to efficiently use limited energy to maintain survival [52]. These findings suggest that nad6 may contribute to environmental adaptation mechanisms in Squamate reptiles [53]. This is crucial for T. scincus to resolve the contradiction between "low food intake and high energy demand" in arid regions and can help it efficiently synthesize ATP under limited resources to maintain energy supply for nocturnal activities (such as predation and enemy avoidance).
Phylogenetic analysis showed that T. scincus and T. keyserlingii clustered into a monophyletic group with high support (BI/ML support > 99%). Moreover, T. przewalskii and T. roborowskii formed another monophyletic group, and T. microlepis was the sister group of the two (Figure 6). This clade division is highly consistent with geographical distribution: the former clade is distributed in the arid regions of Central Asia (including Huocheng, Xinjiang and Iran), and the latter is restricted to the arid regions of northwestern China (Tarim Basin, Turpan Basin), indicating that the geographical isolation effect of the Tian Shan Mountains may play a key role in the species differentiation of the genus Teratoscincus [54]. These results not only confirm the taxonomic status of T. scincus within Sphaerodactylidae but also provide molecular evidence for the "geographical clade" division of the genus Teratoscincus, which can be used to revise the taxonomic system of this genus.
This study has two limitations: first, the phylogenetic analysis only included 10 Sphaerodactylidae species and did not cover all 9 species of the genus Teratoscincus, which may underestimate the deep differentiation within the genus; second, only mitochondrial gene data were used, and nuclear genes were not combined to verify the phylogenetic relationship, making it difficult to exclude the maternal inheritance bias of mitochondrial genes. In the future, it is necessary to supplement Teratoscincus species samples across geographical regions and integrate mitochondrial and nuclear gene data. This will not only clarify the evolutionary history of this genus but also further locate key nuclear gene loci for arid adaptation.
5. Conclusions
Through this study, we successfully determined, assembled, and annotated the complete mitochondrial genome of T. scincus (16,943 bp). A phylogenetic tree was constructed by combining the mitochondrial genomes of nine Sphaerodactylidae species (with E macularius as the outgroup). The results showed the following: The T. scincus mitochondrial genome has a typical vertebrate structure with a significant AT bias (A + T = 55.8%). And Phylogenetic analysis supports that T. scincus and T. keyserlingii, as well as T. przewalskii and T. roborowskii form sister groups, respectively, which confirms the driving effect of geographical isolation imposed by the Tian Shan Mountains on species differentiation within the genus Teratoscincus. This study provides basic data for the taxonomic revision of Sphaerodactylidae and the conservation genetics research of T. scincus.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Midtgaard R. Rep Focus—A Survey of the Reptiles of the World Available online: https://repfocus.com/(accessed on 1 October 2025)
- 2Díaz-Lameiro A.M. Villamil C.I. Gamble T. Pinto B.J. Herrera-Martínez A. Thomas R. Bernstein J.M. Titus-Mcquillan J.E. Nielsen S.V. Agosto-Torres E. A New Species of Sphaerodactylus (Gekkota: Sphaerodactylidae) from the Northwest Limestone Region of Puerto Rico Ichthyol. Herpetol.202211044946510.1643/h 2020123 · doi ↗
- 3Zheng D.Q. Ma R.R. Guo X.G. Li J. Comparative Mitogenomics of Wonder Geckos (Sphaerodactylidae: Teratoscincus Strauch, 1863): Uncovering Evolutionary Insights into Protein-Coding Genes Genes 20251653110.3390/genes 1605053140428353 PMC 12111026 · doi ↗ · pubmed ↗
- 4Uetz P. Freed P. Aguilar R. Reyes F. Kudera J. Hošek J. The Reptile Database Available online: http://www.reptile-database.org/(accessed on 1 October 2025)
- 5Gamble T. A Review of Sex Determining Mechanisms in Geckos (Gekkota: Squamata)Sex. Dev.201048810310.1159/00028957820234154 PMC 2855288 · doi ↗ · pubmed ↗
- 6Badiane A. García-Porta J. Červenka J. Kratochvíl L. Sindaco R. Robinson M.D. Morales H. Mazuch T. Price T. Amat F. Phylogenetic Relationships of Semaphore Geckos (Squamata: Sphaerodactylidae: Pristurus) with an Assessment of the Taxonomy of Pristurus rupestris Zootaxa 20143835335810.11646/zootaxa.3835.1.225081434 · doi ↗ · pubmed ↗
- 7Tamar K. Els J. Kornilios P. Soorae P. Tarroso P. Thanou E. Pereira J. Shah J.N. Elhassan E.E.M. Aguhob J.C. The Demise of a Wonder: Evolutionary History and Conservation Assessments of the Wonder Gecko Teratoscincus keyserlingii (Gekkota, Sphaerodactylidae) in Arabia P Lo S ONE 202116 e 024415010.1371/journal.pone.024415033411750 PMC 7790289 · doi ↗ · pubmed ↗
- 8Clayton D.A. Replication of Animal Mitochondrial DNA Cell 19822869370510.1016/0092-8674(82)90049-66178513 · doi ↗ · pubmed ↗