Genomic Evidence Supporting a One Health Perspective on Staphylococcus aureus Bovine Mastitis
Gabriele Meroni, Valerio Massimo Sora, Giulia Laterza, Alessio Soggiu, Piera Anna Martino, Francesca Zaghen, Luigi Bonizzi, Luciana Colombo, Alfonso Zecconi

TL;DR
This study shows that Staphylococcus aureus in cows and humans have distinct genetic traits, highlighting the importance of a One Health approach to track infections and antibiotic resistance.
Contribution
The study provides genomic evidence of host adaptation and inter-species transmission in Staphylococcus aureus lineages.
Findings
Bovine-adapted S. aureus lineages show reduced human virulence genes and acquired bovine-specific mobile elements.
Animal and human isolates share only ST8, which has different antibiotic resistance profiles.
Bovine isolates generally carry fewer resistance genes than human isolates.
Abstract
Background/Objectives: Staphylococcus aureus is a multifaceted pathogen responsible for diseases in humans and in several animal species, including dairy cows. This study aimed to characterize and compare the genetic diversity, lineage distribution, and antimicrobial resistance profiles of S. aureus isolates from bovine milk with human-derived reference genomes to investigate host adaptation and inter-species transmission. Methods: Genomic analyses were performed on S. aureus isolates from quarter milk samples of dairy cows together with human-derived sequences from public databases. Whole-genome sequencing and multi-locus sequence typing (MLST) were used to determine sequence type (ST) distribution, and the presence of key antibiotic resistance genes and mobile genetic elements (MGEs) was assessed. Comparative genomics was applied to evaluate gene content, phylogenetic relationships,…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
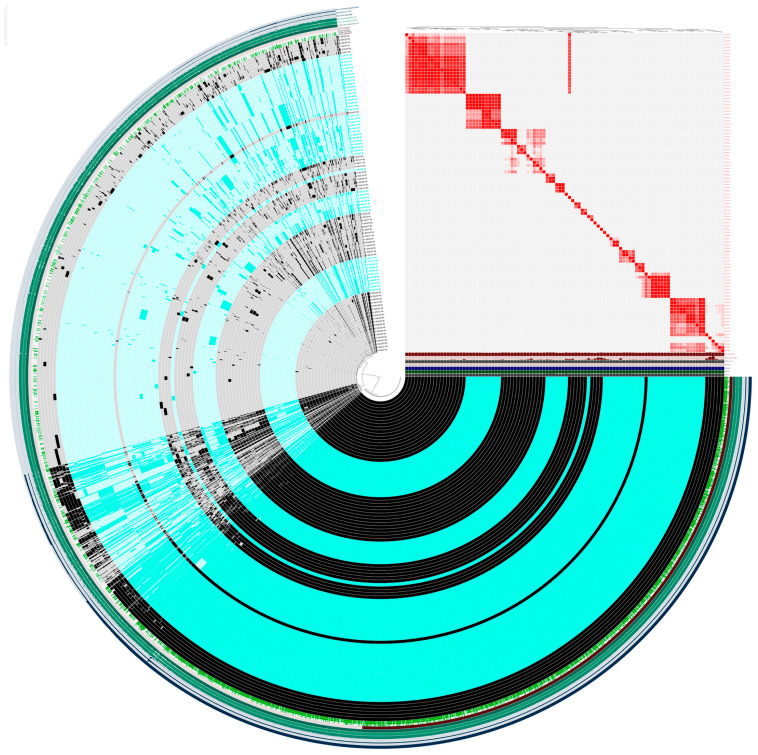 Figure 1
Figure 1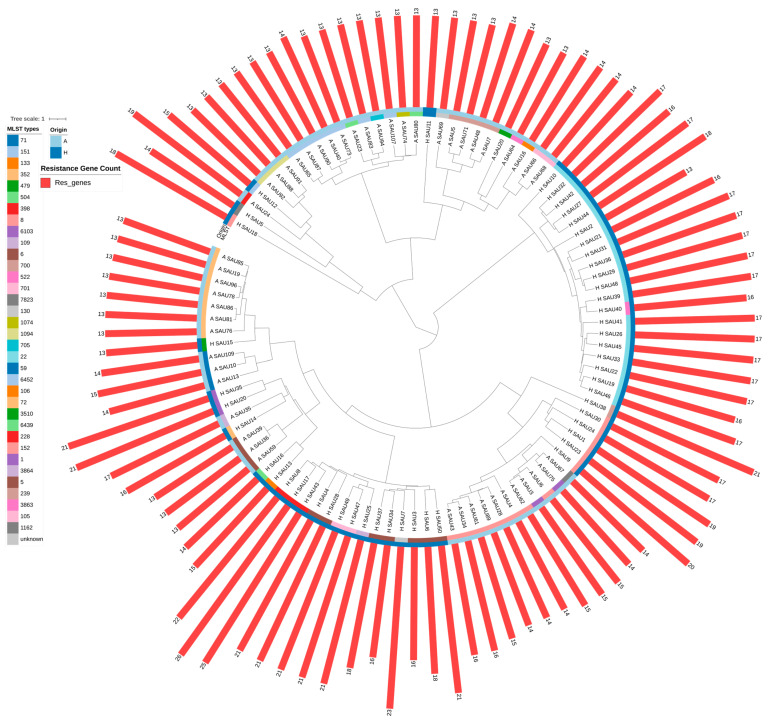 Figure 2
Figure 2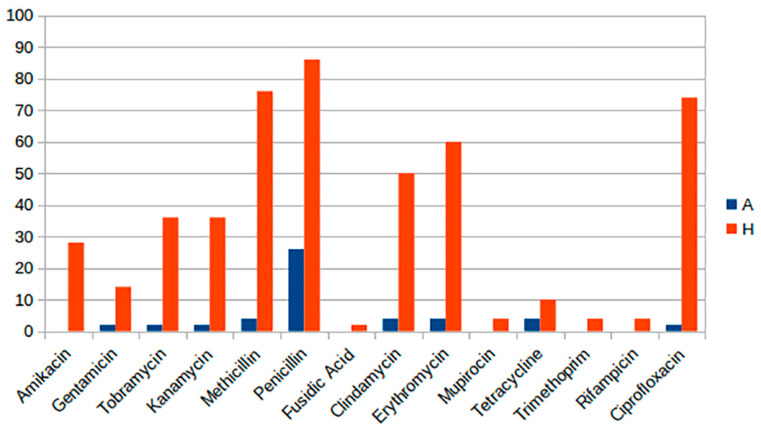 Figure 3
Figure 3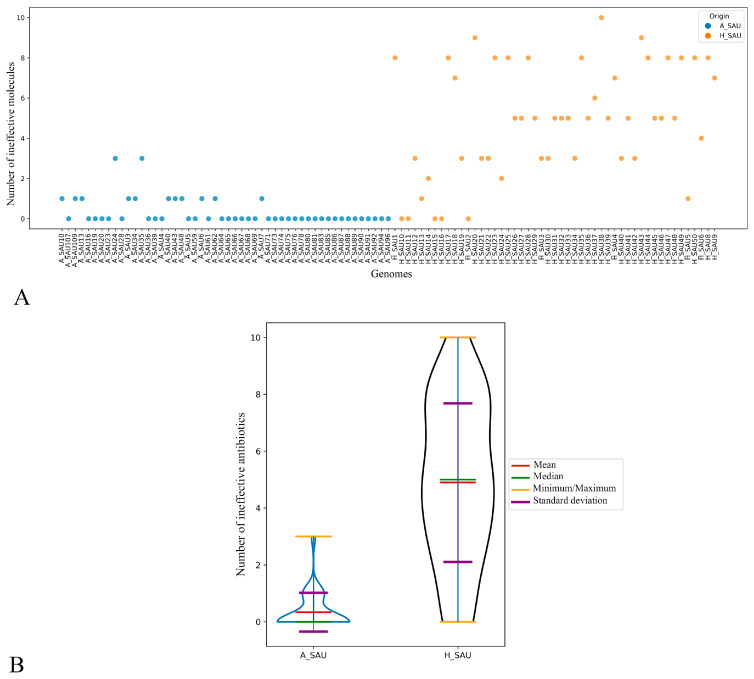 Figure 4
Figure 4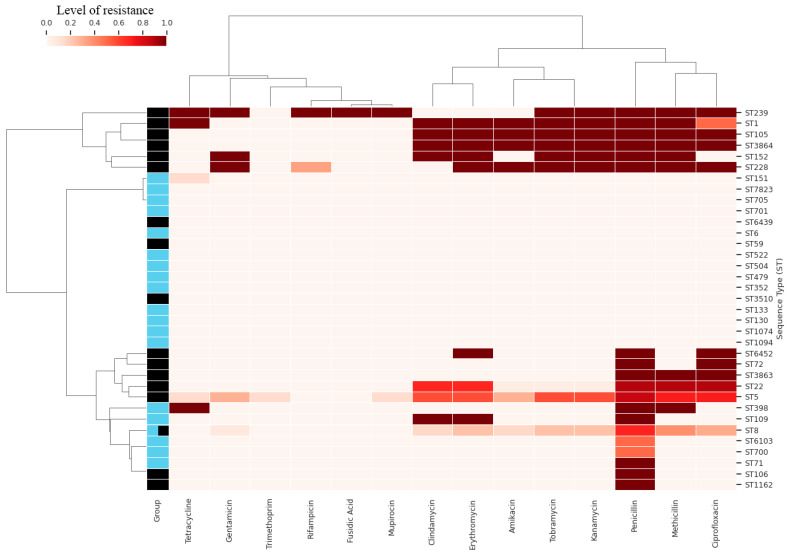 Figure 5
Figure 5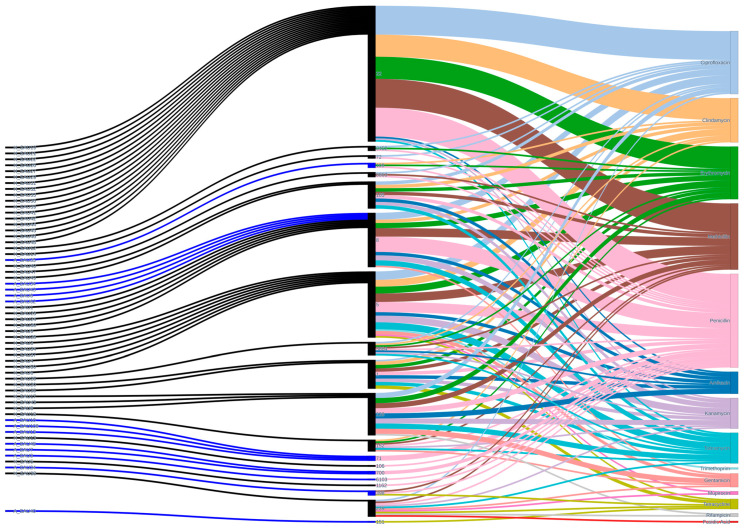 Figure 6
Figure 6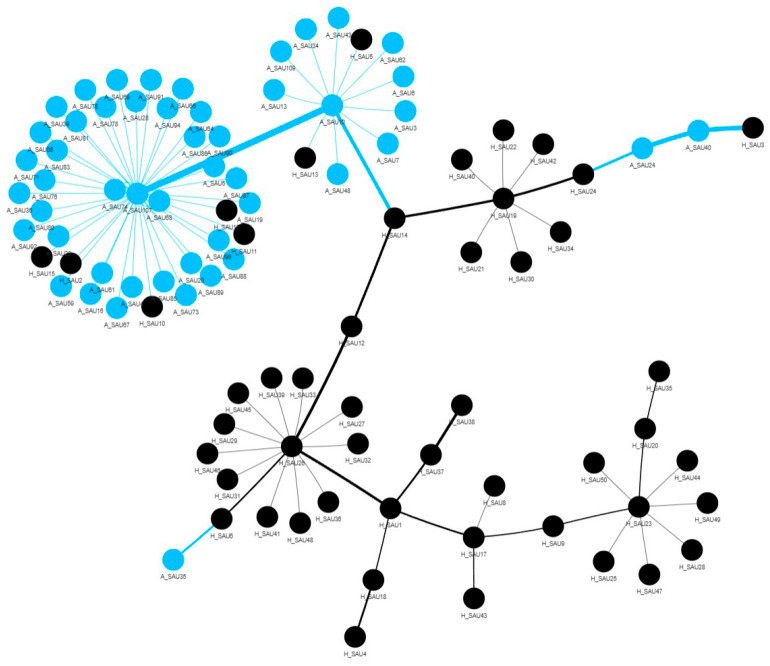 Figure 7
Figure 7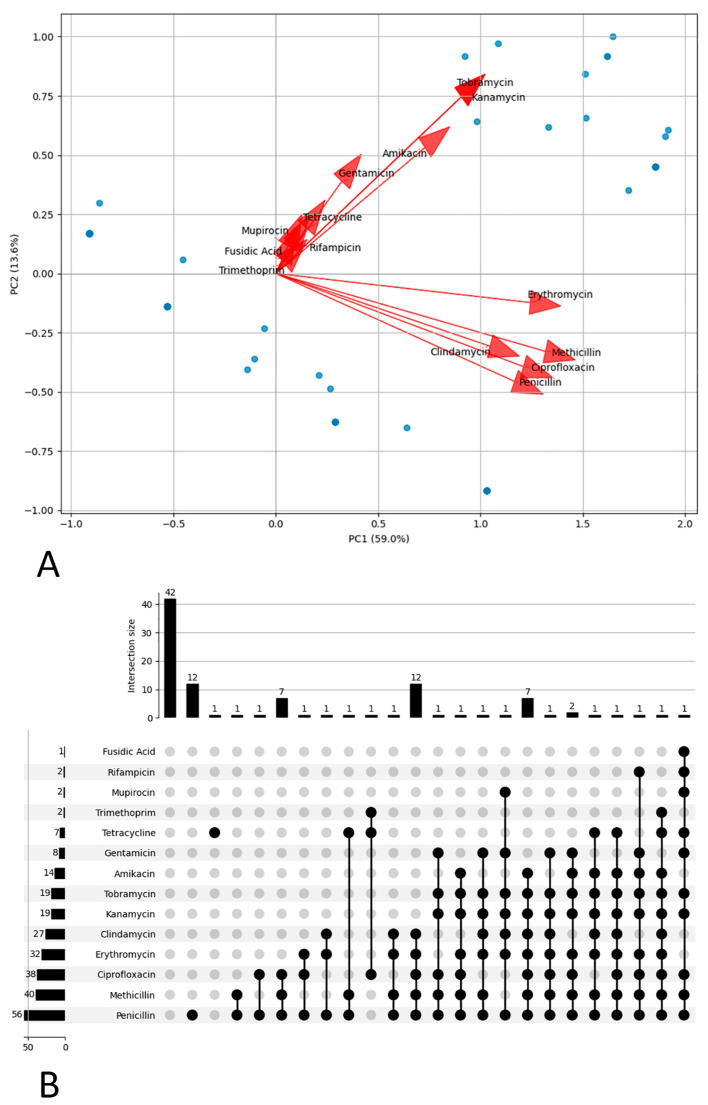 Figure 8
Figure 8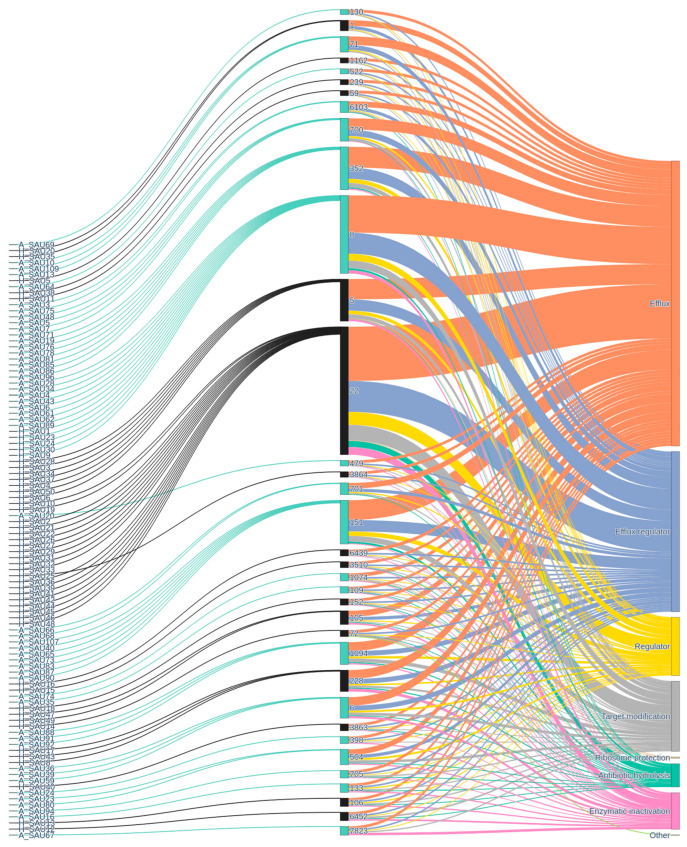 Figure 9
Figure 9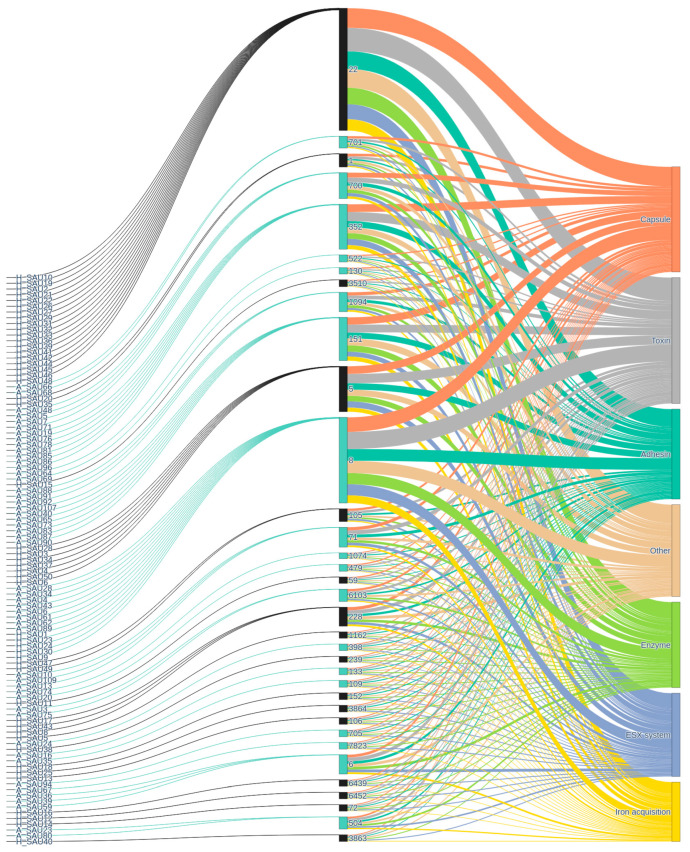 Figure 10
Figure 10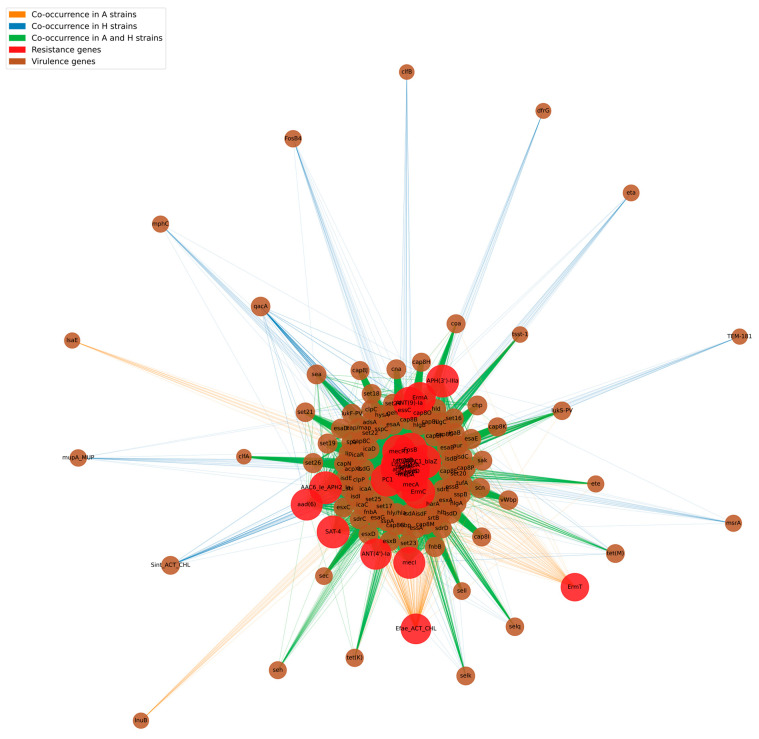 Figure 11
Figure 11- —FEASR—Programma di Sviluppo Rurale 2014–2020 Misura 16.1 project MOOH
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsAntimicrobial Resistance in Staphylococcus · Milk Quality and Mastitis in Dairy Cows · Bacterial biofilms and quorum sensing
1. Introduction
Staphylococcus aureus is known for inducing various diseases in both animals and humans. In humans, it can induce skin infections, pneumonia, and more severe diseases such as endocarditis and toxic shock syndrome [1]. The rise in antibiotic-resistant clones, especially methicillin-resistant Staphylococcus aureus (MRSA), has intensified worries about public health and zoonotic transmission for this pathogen. In dairy cows, it is a principal causative agent of mastitis, resulting in a significant negative economic impact on the dairy industry [2]. Despite the extensive literature on the epidemiology and control of intramammary infections due to S. aureus [3,4,5,6], these infections are still recorded in many countries, and particularly in Italy [7,8,9].
The potential role of milk as a source of food-poisoning due to S. aureus enterotoxins is well known [10], as is its role in the case of livestock-associated methicillin resistance [11]. However, the potential zoonotic role of S. aureus commonly associated with intramammary infections in dairy cows is still unclear. This pathogen has a vast array of virulence factors and antimicrobial resistance genes [12,13,14,15,16,17] and its genetic apparatus suggests that the potential zoonotic capabilities should not be ignored.
The genetic diversity of S. aureus is evident in its multi-locus sequence typing (MLST), which classifies the bacterium into various sequence types (STs) and clonal complexes (CCs) [18]. Several STs are notably shared across humans and animals, revealing a complex global health interaction and making a One Health approach relevant. ST398 is primarily linked to livestock, mainly pigs, although it has also been detected in humans, particularly those with close contact to these animals [19]. Likewise, ST5 and ST6 have been identified in both meat products and human clinical isolates, indicating that foodborne transmission pathways may contribute to the epidemiology of S. aureus infections [20]. Besides cattle, wildlife has also been involved in the transmission dynamics of S. aureus with ST49 and ST581 being the main STs sharing genetic similarities with human isolates [21,22]. The existence of these shared STs prompts enquiries into the biological mechanisms that enable their persistence among various species. Bovine-adapted S. aureus lineages include CC97, CC133, and CC151 while human-adapted lineages include CC1, CC5, CC8, CC30, and CC45 [5]. S. aureus host spillover followed by adaptation to a new host is typically associated with the acquisition of a new set of genes specific for survival in the new host losing virulence to the former host species [23]. Recent comparative genomic reports indicate that bovine-adapted strains swiftly discard genes associated with human infections, thereby likely enhancing their fitness in bovine hosts [5,23,24]. Moreover, cows serve as a reservoir for antimicrobial-resistant (AMR) S. aureus, which could be transmitted to people and persist within them [12,13]. Few studies have tried to understand this association by correlating AMR and lineage. Few STs are known to be positive for blaZ or mecA among the S. aureus CC97 isolated from cows, which comprises many STs (ST97, ST115, and ST352). Moreover, ST5 (CC5), ST8 (CC8), and their variants are human-adapted lineages that have been isolated from cows and were positive for blaZ and mecA [25,26,27].
Despite its significance in human and veterinary medicine, research is scarce on the gene profile, strain-to-strain relationships, disease–host relationships, and host–pathogen interactions of S. aureus. We implemented genome-wide analyses in the present study to gain a more thorough understanding of these issues by comparing S. aureus strains isolated from the milk samples of dairy cows and human-derived sequences from public databases.
2. Results
2.1. Genomic Features of the Analyzed Strains
We performed Nanopore Whole Genome Sequencing on 50 S. aureus strains isolated from bovine milk. All the sequences were deposited in NCBI under the Bioproject PRJNA1243330. Table 1 summarizes the main descriptors of the sequenced strains. The distribution of agr types, SCCmec types, and sequence types (STs) among human (H_) and animal (A_) isolates reveals distinct population structures characterized by specific host-associated predominance. Regarding the agr locus, human isolates are predominantly agr group 1 (66%), followed by group 2 (30%), and group 3 (4%). Animal isolates show a similar pattern with slightly less dominance of group 1 (64%), group 2 (26%), and a higher proportion of group 3 (10%). The SCCmec typing, highlights the absence of methicillin resistant strains in the A_ cohort (98%), with the detection of a single strain found positive for mecA gene and clustered as SCCmec type V:Vc. In contrast 36% of H_ strains carried SCCmec IV:IVj type, 12% II:IIa and IV (other subtypes found comprise SCCmec types IV:IVa (6%), I:Ia (6%), as well as IV, V, and III (each 2%)).
Sequence type analysis further emphasizes the circulation of distinct clones in humans and animals. ST22, a lineage globally recognized as a hospital-associated MRSA strain linked to epidemic spread and high virulence, is the most represented ST in H_ strains (38%). Other human-associated STs include ST5 (14%), ST8 (10%), and additional less frequent STs such as 228 (6%), 105 (4%), 1 (4%). In A_ isolates, ST8 is the most frequent (16%), closely followed by ST352 and ST151 (14%), ST700 (8%), and other STs such as 71, 1094, 6, 6103, 701, 504.
2.2. Pangenome Analysis
Figure 1 (see also Supplemental Figures S1–S4) illustrates the pangenome generated with the ANVI’O pipeline.
The pangenome comprised 7200 gene clusters, including 1729 core genes (found in at least 99% of strains), 157 soft-core genes (present in 95–99% of strains), 1352 shell genes (in 15–95% of strain), and 3962 cloud genes found in less than 15% of strains. The dominance of cloud genes, which represent more than half of the pangenome, together with a sizeable shell fraction, is consistent with an open pangenome and extensive genomic flexibility. The separation between core and soft-core components highlights a conserved backbone of housekeeping and essential functions, accompanied by a subset of quasi-essential genes whose incomplete distribution suggests ongoing gene gain and loss dynamics. This soft-core fraction likely reflects adaptation to specific ecological contexts or hosts, where certain functions provide a selective advantage without being strictly required across all lineages.
The accessory genome (shell and cloud) accounts for the majority of gene clusters and underscores marked variability among strains, with many genes present in only a limited number of strains, thus indicates a high genomic plasticity and suggests that horizontal gene transfer and niche-specific selection are important drivers of species evolution.
Clustering patterns based on gene presence–absence and average nucleotide identity (ANI) revealed groups of highly similar genomes, often corresponding to strains of shared animal or human origin. These clusters indicate that host-associated lineages tend to retain more similar gene clusters, supporting the contribution of host adaptation to the shaping of the accessory genome.
The combination of a stable core and a highly dynamic accessory genome likely facilitates persistence in different hosts and environments, and may underpin differences in virulence, colonization ability, or antimicrobial resistance potential between animal- and human-derived strains.
2.3. Phylogeny
Whole genome phylogenetic reconstruction (Figure 2) revealed substantial genetic diversity, with animal- and human-derived strains distributed across distinct branches and, in several cases, clustering within shared clades. A complex evolutionary pattern is indicated by the existence of both host-specific lineages and mixed clades. This pattern is marked by separate host-associated divergence in some lineages, while other lineages exhibit indications of genetic mixing or recent zoonotic trade.
A difference in accessory gene acquisition emerged between the two populations; human-derived isolates harbored significantly more resistance genes (mean 17.9 per genome) compared to animal-derived strains (mean 13.8 per genome; Welch’s t-test, p < 0.0001). This pattern suggests that human-associated lineages have accumulated antimicrobial resistance determinants, consistent with exposure to antimicrobial selective pressure in clinical and community settings. Conversely, animal-derived strains displayed a higher mean count of virulence-associated genes (77.02 vs. 74.64; Welch’s t-test, p = 0.0043), suggesting that preservation of a broader virulence factor repertoire may be maintained through ecological or infection-related selection in animal hosts.
These results indicate that whilst a conserved phylogenetic backbone links the species, differential acquisition and dissemination of resistance and virulence genes reflects distinct selective pressures ongoing in animal and human hosts, with implications for pathogenic potential and treatment challenges in each population.
2.4. Antibiotic Resistance of S. aureus in Human (H_SAU) and Animal (A_SAU) Strains
Figure 3 summarizes the predicted phenotypic antibiotic resistance detected in the two groups (H_SAU and A_SAU).
Predicted antibiotic resistance distribution reveals substantial disparities, with increased resistance in H_SAU across all examined antibiotic classes. Specifically, amikacin resistance is absent in animal strains, whereas it is present in 28% of human strains. Resistance to aminoglycosides (gentamicin, tobramycin, and kanamycin) in human strains is 14%, 36%, and 36%, respectively, although in animal strains, it ranges from 2% to 4%. Significant disparities are noted with methicillin (4% A vs. 76% H), penicillin (26% A vs. 86% H), erythromycin (4% A vs. 60% H), and ciprofloxacin (2% A vs. 74% H). Certain antibiotics, including fusidic acid, mupirocin, trimethoprim, and rifampicin, exhibit no resistance in animal strains, but human strains demonstrate resistance rates ranging from 2% to 4%.
In the scatter plot (Figure 4A), most of the animal samples did not show any resistance or exhibit resistance to a maximum of two antibiotics. The plot shows a clear separation between animal- and human-derived strains, with animal isolates (n = 50) exhibiting a very low resistance burden (mean 0.34 resistant antibiotics per strain; median 0; range 0–3) and human isolates (n = 50) displaying a substantially higher load (mean 5 resistant antibiotics per strain; median 5; range 0–10). This pattern indicates that multiple resistance phenotypes are largely confined to the human population, where individual isolates frequently combine resistance to several classes, including beta-lactams (methicillin and penicillin), aminoglycosides (amikacin, gentamicin, tobramycin, kanamycin), macrolides and lincosamides (erythromycin, clindamycin), fluoroquinolones (ciprofloxacin), and occasionally rifampicin, mupirocin or tetracycline, whereas animal isolates are typically fully susceptible or resistant only to penicillin, with sporadic resistance to tetracycline. No animal isolates can be defined as MRSA.
The violin plot (Figure 4B) represents the distribution of the number of resistances for each group. Animal strains show a highly compressed distribution, with a median of 0 resistant antibiotics per isolate (interquartile range [IQR] 0–0; range 0–3), whereas human strains exhibit a much broader and right-shifted distribution, with a median of 5 resistances per isolate (IQR 2–7; range 0–10). This difference in distributions is statistically significant (e.g., Welch’s t-test or a non-parametric alternative; p < 0.0001), reflecting both the higher central tendency and the greater dispersion of resistance counts among human isolates compared with the tightly clustered, mostly fully susceptible profiles observed in animal isolates.
The heatmap (Figure 5) clusters the STs of the two groups based on the level of antibiotic resistance. Some STs assigned to the A_SAU group show no resistance, while most of the human samples are resistant to multiple antibiotics (as shown in the previous figures). The only ST common to the two groups is ST8, which however shows different levels of resistance between the two groups. Indeed, ST8 from animal isolates carried resistance determinants that were not detected in the human ST8 isolates, specifically PC1_blaZ, AAC6_Ie_APH2_Ia, and ermC. Conversely, resistance genes identified exclusively in human ST8 isolates were tet(K), aad(6), SAT-4, and APH(3′)-IIIa. These differences may be useful to identify the source of the ST8 isolates (e.g., in an outbreak epidemiological investigation).
Among the antibiotics analyzed, penicillin is the one towards which most STs show resistance, followed by methicillin, ciprofloxacin, erythromycin and clindamycin (Figure 6).
The Minimum Spanning Tree (MST, Figure 7) highlights clusters of related strains based on the number of antibiotic resistances genes (ARGs). Two main clusters can be observed: one in the top left, composed almost exclusively of animal samples, and one in the bottom, predominantly human. The human cluster appears more fragmented, with several sub-clusters. This could indicate a greater diversity of genetic resistance profiles in the human STs, perhaps due to different selective pressure. The presence of nodes of opposite color in the two clusters (A_ or H_), suggest that STs that share similar resistance profiles in both animal and human samples. These nodes are particularly interesting because they could represent interspecific transmission events or the emergence of clones adapted to both hosts.
The Principal Component Analysis (PCA) results (Figure 8A) show that the two principal components together explain approximately 72.64% of the total variance in the resistance gene data, with PC1 alone accounting for 59% and PC2 for 13.64%. The PC1 principal component represents the main axis of variation, while PC2 adds a complementary dimension. Analyzing the contributions of the ARGs to the principal components, it is observed that PC1 is characterized by methicillin (0.439), erythromycin (0.414), ciprofloxacin (0.403), clindamycin (0.349), penicillin (0.389) and kanamycin/tobramycin (approximately 0.30) ARGs. These results indicate that variability in resistance to these antibiotics is the main factor distinguishing the strains along PC1. PC2, with positive contributions from kanamycin, tobramycin, amikacin and gentamicin ARGs, may reflect a secondary dimension of variation.
The analysis of ARGs combinations by means of the UpSet plot (Figure 8B) allows to monitor the evolution of resistance profiles and to detect the appearance of new multiresistance patterns. The results show that the most frequent combinations involve several ARGs: amikacin, tobramycin, kanamycin, methicillin, penicillin, clindamycin, erythromycin and ciprofloxacin. The recurrence of these combinations in many strains suggests the presence of co-selected resistance mechanisms. Aminoglycosides (amikacin, tobramycin and kanamycin) ARGs are frequently associated, which is consistent with the presence of aminoglycoside-modifying enzymes (AMEs) that confer cross-resistance to multiple molecules of this class. The concomitant presence of resistance to beta-lactams (methicillin and penicillin) indicates the spread of strains by multiple mechanisms, such as beta-lactamase production or alterations of the PBP (penicillin-binding protein) target.
Increased resistance in human-derived isolates, particularly to first-line agents such as penicillin, methicillin, erythromycin, clindamycin and ciprofloxacin, has direct implications for empirical therapy and the risk of treatment failure in clinical settings. The lower resistance burden in bovine isolates indicates that dairy herds may act as reservoirs of resistant S. aureus, with potential spillover into humans through occupational exposure or the food chain, reinforcing the need for coordinated antimicrobial stewardship under a One Health framework.
2.5. Resistance Genes and Virulence Factors
Figure 9 shows the relationships among the bacterial strains analyzed, their STs, the ARGs identified, and the associated molecular mechanisms. The first level, consisting of the strains, shows significant diversity, with numerous isolates distributed across different STs, suggesting considerable genetic heterogeneity and the possible presence of clones. The strain → ST transition highlights some particularly well-represented STs, which act as hubs in the network (i.e., ST22, ST8, ST352, ST151). This pattern may reflect the presence of clones that have spread across different contexts, probably due to selective advantages conferred by resistance or virulence. Some STs are associated with a broad spectrum of ARGs, while others show a more limited carriage (ST7823). The presence of multiple ARGs in some STs suggests the co-localization of mobile genetic elements, such as plasmids or transposons, which facilitate their horizontal spread. The last level, resistance gene → mechanism, represents the functional categorization of the identified genes. Here, some dominant mechanisms clearly emerge, such as enzymatic modification of the antibiotic, target modification, active efflux and target protection.
Figure 10 shows the distribution and association of virulence factors in the strains analyzed. Starting from the isolates, the graph shows a structure similar to that observed for resistance, with numerous samples converging on a relatively limited number of STs. This pattern suggests that, even for virulence, there are dominant clones that serve as the main carriers of certain virulence factors within the population. The strain → ST transition again highlights some ST, which bring together numerous strains (ST22, ST8), suggesting a complex clonal structure and the possible presence of hypervirulent clones. Some STs are associated with a broad spectrum of virulence factors, which may facilitate adaptation to different hosts or ecological niches. Other STs, on the other hand, show a “specialization” towards certain categories (penicillin and tetracycline ARGs), indicating a possible narrower ecological niche or infection strategy (ST109, ST152, ST106). The presence of numerous connections between STs and virulence categories suggests that genetic plasticity affects not only resistance but also pathogenic potential. The figure shows that some categories of virulence factors are particularly well represented, such as adhesins and toxins, while others, such as immune evasion factors or degradative enzymes, are less frequent or more specific to certain STs.
To better determine the relationship between resistance and virulence genes, a network analysis was performed focusing on co-occurrence (Figure 11). The arcs connecting the nodes are colored orange if the co-occurrence is exclusive to the A_ strains, purple if exclusive to the H_ strains, and lilac if both groups share the co-occurrence. The size of the nodes is proportional to the degree of connection (i.e., the number of genes with which each resistance or virulence gene co-occurs), while the thickness of the arcs reflects the frequency of co-occurrence. The network is characterized by most resistance genes connected to many virulence factors, and vice versa. This suggests that, in the strains analyzed, resistance genes are not isolated events but are accompanied by several virulence genes, highlighting the genetic plasticity of S. aureus. Most co-occurrences are shared between the two groups of strains (lilac), while only a minority of arcs are exclusive to strains A_ (orange) or H_ (purple). This suggests that the structure of the co-occurrence network is largely conserved between the two groups.
3. Discussion
Staphylococcus aureus has emerged as a paradigm of host adaptation, clonal evolution, and global distribution among pathogens with zoonotic and anthropozoonotic potential. The results of our comparative genomic analysis of staphylococcal strains isolated from milk samples and human clinical sources expand current knowledge on host-specialized lineages, genetic diversification, and the mechanisms underlying host jumps. We critically discuss the implications of these models, referring to comparative genomics, population-level investigations, phylogenetics, antimicrobial resistance (AMR), and virulence traits.
The clonal diversity of S. aureus in both humans and cattle reflects a global trend that determines the epidemiology in each host. Our data, in line with other investigations, confirm that CC97, CC133, and CC151 are the main complexes adapted to cattle, consistent with the results of molecular and genomic typing studies demonstrating their prevalence in dairy herds in Europe, North America, Asia, and Africa [1,28,29]. This is reflected in human populations by dominant clonal complexes such as CC1, CC5, CC8, CC30, and CC45 [29,30]. Several studies report the presence of “human” lineages (particularly CC8 and CC5) in cattle and, conversely, the detection of “zootechnical” lineages such as ST398 in human carriers, especially among individuals who work closely with livestock [29,31,32,33]. Our results support these findings, with evidence of sporadic host shifts and complex patterns of clonal overlap, although true sustained host adaptation appears relatively rare for most lineages. Bovine-adapted lineages are characterized by the acquisition of specific phages and pathogenicity traits, supporting genetic events that facilitate adaptation to the bovine mammary niche. Human-adapted strains often contain prophages that modify β-hemolysin and encode immune evasion factors particular to humans [34]. It is crucial to acknowledge that transitions from human to bovine, exemplified by CC8, involve the loss of human-specific phage elements concomitant with the acquisition of novel SCC cassettes, occasionally encoding proteins featuring the new LPXTG motif. This phenomenon appears to facilitate rapid adaptation, as evidenced by our research and by comparative microarray-based genomics conducted in China and Switzerland, respectively [32,33,35].
Principal component analysis (PCA), and minimal spanning tree (MST) are examples of multivariate statistical studies that help us understand the population structure. PCA shows that resistance to important clinical antibiotics like methicillin, erythromycin, and ciprofloxacin is the main factor that separates strains. Resistance to macrolides and lincosamides (erythromycin and clindamycin) is prevalent in the most common combinations, probably mediated by ribosomal target modifications or efflux pumps. Co-resistance of beta-lactams and aminoglycosides is frequently associated with plasmids with multiple gene cassettes, while resistance to macrolides and lincosamides may arise from erm or mef genes, often co-localized with other resistance genes. Moreover, the influence of beta-lactam antibiotics (methicillin and penicillin) and macrolides (erythromycin and clindamycin) ARGs on PC1 underlines the importance of closely monitoring resistance to these classes, which represent the first choice for empirical therapy. In addition, the relevant contribution of ciprofloxacin highlights the need to also consider resistance to fluoroquinolones [36,37]. The MST shows distinct clusters that are mostly based on the host’s origin, but there are also important bridging nodes that signal genetic overlaps and possibly cross-transmission or shared environmental reservoirs. This supports the idea that pathogens’ evolution and ecology are connected in ways that make it harder to develop controls measures to manage them separately. Therefore, we need to think about diversity in pathogen genomics in terms of ecology and evolution.
Recent studies that used global phylogenetic reconstruction show that clonal complexes switch hosts at different rates and in different ways. The dairy bovine population acts more like a “sink” than a “source” for host hops, save for certain lineages like CC425 [38]. Modeling shows that ecological factors including farming practices, antibiotic use, and herd management, as well as microevolutionary adaptation, all affect how populations of cattle and people change over time [38,39,40]. In our strains, the presence of compact clades consisting exclusively of animal or human strains suggests that, despite the possibility of transmission between species, there are still selective or ecological barriers that favor the specialization of certain evolutionary lines. The presence of mixed clades, on the other hand, suggests that in some circumstances these barriers can be overcome, allowing the transmission of strains between animals and humans.
S. aureus can swap hosts and spread from animals to people because it has a lot of genetic variation and can adapt to new environments. Finding different but overlapping clonal populations shows that some strains may be better suited to certain hosts, but ecological barriers are not always strong enough to stop genetic exchange and adaptation. This ability to adapt is made worse by the continual antibiotic selective pressures that cause multi-drug resistance phenotypes to appear, especially in the clinical setting for humans. The fact that animal strains have a lower but still significant level of resistance means that there is still a risk of reservoirs existing in livestock populations, which could be sources of resistant diseases that could spread to people. Resistance genes such as tet(38), dfrC, norA, norC, sepA, mepA, mgrA, sdrM, arlS, arlR, mepR, kdpD, LmrS are associated with a wide range of virulence factors (set22, cap8G, fnbB, clpC, hly/hla, ebp, esaD, sspA, icaR, etc.). This breadth of co-occurrences suggests that most strains possess multiple resistance and virulence determinants simultaneously. The central structure of the network, in which resistance nodes are most represented (such as tet(38), norA, norC, dfrC, sepA, mepA, mgrA), shows a high degree of connection, indicating that these genes play a key role in the bacterial population analyzed. They could represent “core” genes, whose presence is almost ubiquitous, and which act as a platform for the acquisition or co-localization of additional virulence genes. Because of the complicated genetic networks and the fact that resistance and virulence genes often happen at the same time, interventions that only look at resistance may miss important parts of how pathogens spread and how they cause disease.
However, this study has some limitations that should be considered when interpreting the results. First, the sample size and restriction to dairy herds from a single geographic region limit the generalizability of the findings to other production systems and epidemiological contexts. Second, human comparator genomes were selected from Italian public database entries and lack detailed and harmonized metadata on clinical presentation, which constrains inferences on directionality and frequency of cross-species transmission. Finally, the analysis is based on genomic prediction of resistance and virulence rather than systematic phenotypic characterization, so functional consequences of specific gene constellations should be confirmed in future experimental and longitudinal studies.
These results show how important it is to have integrated surveillance systems that can follow the evolution of resistance in S. aureus and the ways it spreads across both human and animal populations. This supports the One Health concept for addressing antibiotic resistance and zoonotic diseases caused by S. aureus. Reducing AMR requires coordinated efforts of human health care, veterinary medicine, farming, and environmental management. To stop the emergence of resistant and virulent S. aureus strains, it is important to use antibiotics wisely in both clinical and agricultural contexts, improve genomic surveillance, and take targeted steps to prevent infections. Further studies on the molecular mechanisms that allow hosts to adapt and pathogens to become resistant could aid in the development of new interventions to reduce the spread of this pathogen.
4. Materials and Methods
4.1. Bacterial Isolation and Identification
Isolates were collected in the period January 2024–May 2025 from the dairy herds in Lombardy region applying a mastitis control program and delivering samples to ARAL (Associazione Regionale Allevatori Lombardia) laboratories. Only S. aureus isolated from quarter milk samples of cows with subclinical mastitis were considered. Diagnosis was performed following current diagnostic protocols applied in ARAL laboratories and following standardized procedures [4,41]. A positive S. aureus mastitis was defined when the bacteria was found in pure culture or when no more than two different bacteria species were observed. For each herd, a single isolate was considered. When more than one cow was positive within a herd, the selection criteria were: presence of a pure culture and highest somatic cell count.
The bacteriological analysis of the single-quarter milk samples was conducted in accordance with the guidelines established by the National Mastitis Council (NMC). Milk samples were inoculated onto Tryptic Soy Agar (TSA, Microbiol, Uta, Cagliari, Italy) supplemented with 5% defibrinated sheep blood, and Baird-Parker agar plates (Thermo Fisher, Milan, Italy), subsequently incubated aerobically at 37 °C for 24 h. Colonies presumptively identified as staphylococci based on golden-yellow pigmentation and hemolytic activity, underwent Gram staining. After staining, confirmatory tests include catalase, coagulase and biochemical tests performed on Vitek^®^ system (BioMerieux, Lyon, France).
4.2. DNA Extraction
Isolated colonies from fresh overnight culture of S. aureus on TSA agar were resuspended in 500 μL of PBS (Phosphate-Buffered Saline, Euroclone, Milan, Italy) and centrifuged at 10,000× g for 1 min at room temperature. DNA was extracted using the Quick-DNA™ HMW MagBead kit (Zymo Research, Irvine, CA, USA) in accordance with the manufacturer’s guidelines.
The quantity and quality of the extracted DNA were evaluated using UV-Vis spectrophotometry with the NanoReady Touch Micro Volume Reader (Aurogene, Rome, Italy), confirming that the A_260_/A_280_ and A_260_/A_230_ ratios fell within the range of 1.8 to 2, respectively.
4.3. Sequencing and Bioinformatics Analyses
Genomic libraries were constructed using 200 ng of input DNA per sample, which underwent tagmentation using transposase with the Rapid Barcoding Sequencing kit (SQK-RBK114.96, Oxford Nanopore Technologies, Oxford, UK). Subsequently, twelve strains were loaded on a single flowcell (FLO-MIN114, version R10.4.1) and sequenced using the third-generation MinION Mk1C sequencer (Oxford Nanopore Technologies, Oxford, UK) for up to 72 h.
Dorado (v0.8.2, [42]) was used for basecalling, adapter trimming, and demultiplexing. Summary data about the quality and amount of reads were generated using NanoPlot (v1.44.0, [43]). Filtration was conducted using FiltLong (version 0.2.1, [44]). Subsequently, the genomes were de novo assembled using Flye (v2.8.1-b1676, [45]), and the resulting contigs were polished and corrected via Medaka (v2.0.1, [46]). The assessment of genomic completeness and contamination was conducted using CheckM2 (v1.2.4, [47]). Structural and functional genome annotation was performed using the NCBI Prokaryotic Genome Annotation Pipeline (PGAP, [48]), which identifies and annotates coding sequences, rRNAs, and tRNAs. Pan-genome analyses, including identification of core and accessory genes and construction of the pangenome matrix, were carried out with PGAP2 [49] and complementary tools such as Anvi’o [50]. Average nucleotide identity was assessed using ANIClustermap tool [51] and the phylogenetic tree was visualized with iTOL (v.6) [52].
The genomes of the isolated bacteria were compared with those in the database (Pathogen Detection, GenBank, https://www.ncbi.nlm.nih.gov/pathogens/ accessed on December 2024) to analyze pathogenic features, including clonality and the presence of antibiotic resistance genes. Fifty genomes of S. aureus, obtained from human isolates, were randomly chosen from samples collected in Italy (Supplemental Table S1).
4.4. Statistical Analysis
A Chi-squared test was used for categorical variables (e.g., resistant vs. susceptible strains, animal vs. human), and Welch’s t-test for continuous variables (e.g., number of resistance or virulence genes per isolate); a p-value < 0.05 was deemed statistically significant for all analyses. Analyses (Chi-squared test and inspection of variable distributions by histograms, Q–Q plots, and boxplots prior to parametric testing) were originally performed in Excel™ (Microsoft Corp., Redmond, WA, USA).
Principal component analysis (PCA) was performed on the antibiotic resistance profiles using scikit-learn’s PCA implementation in Python (v3.13), retaining two principal components. Each isolate was represented by its numeric resistance values across antibiotics; features were centered (default behavior of PCA in scikit-learn) but not scaled to unit variance, and principal components were obtained from the eigendecomposition of the covariance matrix, with loadings computed as component coefficients multiplied by the square root of the explained variance and visualized in a biplot.
For clustering, pairwise dissimilarities between isolates were computed on binary (0/1) resistance profiles using the Jaccard distance, and agglomerative hierarchical clustering with average linkage was applied to generate a dendrogram of isolates. The same Jaccard distance matrix was used to construct a complete weighted graph in Python (Linux environment), from which a minimum spanning tree (MST) was obtained using a standard minimum-spanning-tree algorithm and visualized with node colors indicating isolate origin.
5. Conclusions
The results of this study are broadly consistent with current knowledge on Staphylococcus aureus, while providing additional resolution on host association and genomic diversity. The data indicate strong, although not absolute, host adaptation, together with a dynamic capacity for host switching mediated by mobile genetic elements (MGEs) and patterns of antimicrobial resistance. The analyses further identify lineage-specific adaptation events and reveal complex transmission pathways involving both animal and human populations, underscoring the importance of integrated, genomics-informed surveillance.
Despite these advances, important gaps remain in understanding the relationship between genomic variation and pathogenic potential. In particular, the mechanisms that constrain or facilitate host jumps, as well as the ecological determinants that shape local and global distribution patterns, are still only partially resolved. Addressing these gaps will require a comprehensive approach that integrates comparative genomics, population-level epidemiology, experimental studies of pathogenesis, and multi-host ecological investigations. Such an integrated framework is essential to improve the control of a pathogen that exhibits adaptable and dynamic host interactions in both bovine and human populations, and to inform coordinated monitoring systems that are globally connected, genomics-based, and explicitly ecology-aware.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Campos B. Pickering A.C. Rocha L.S. Aguilar A.P. Fabres-Klein M.H. de Oliveira Mendes T.A. Fitzgerald J.R. de Oliveira Barros Ribon A. Diversity and Pathogenesis of Staphylococcus aureus from Bovine Mastitis: Current Understanding and Future Perspectives BMC Vet. Res.20221811510.1186/s 12917-022-03197-535331225 PMC 8944054 · doi ↗ · pubmed ↗
- 2HeikkiläA.-M. Liski E. PyöräläS. Taponen S. Pathogen-Specific Production Losses in Bovine Mastitis J. Dairy Sci.20181019493950410.3168/jds.2018-1482430122416 · doi ↗ · pubmed ↗
- 3Zecconi A. Piccinini R. Fox L.K. Epidemiologic Study of Intramammary Infections with Staphylococcus aureus during a Control Program in Nine Commercial Dairy Herds J. Am. Vet. Med. Assoc.200322368468810.2460/javma.2003.223.68412959389 · doi ↗ · pubmed ↗
- 4Zecconi A. Calvinho L.F. Fox K.L. Staphylococcus aureus Intramammary Infections FIL/IDF Brussels, Belgium 2006 Volume 408136
- 5Park S. Ronholm J. Staphylococcus aureus in Agriculture: Lessons in Evolution from a Multispecies Pathogen Clin. Microbiol. Rev.202134 e 00182-2010.1128/CMR.00182-2033568553 PMC 7950364 · doi ↗ · pubmed ↗
- 6Dufour S. Dohoo I.R. Barkema H.W. Descôteaux L. Devries T.J. Reyher K.K. Roy J.-P. Scholl D.T. Manageable Risk Factors Associated with the Lactational Incidence, Elimination, and Prevalence of Staphylococcus aureus Intramammary Infections in Dairy Cows J. Dairy Sci.2012951283130010.3168/jds.2011-471122365211 · doi ↗ · pubmed ↗
- 7Zaghen F. Sora V.M. Zanirato G. Zecconi A. From One Heath to One Sustainability: The Role of Contagious Mastitis Pathogens in Decreasing the Dairy Herd Sustainability Pathogens 20241391410.3390/pathogens 1310091439452785 PMC 11510415 · doi ↗ · pubmed ↗
- 8Sora V.M. Panseri S. Nobile M. Di Cesare F. Meroni G. Chiesa L.M. Zecconi A. Milk Quality and Safety in a One Health Perspective: Results of a Prevalence Study on Dairy Herds in Lombardy (Italy)Life 20221278610.3390/life 1206078635743817 PMC 9225654 · doi ↗ · pubmed ↗