Maternal Embryonic Leucine Zipper Kinase (MELK) in Cancer: Biological Functions, Therapeutic Potential, and Controversies
Alaeddin M. Alzeer, Saad Al-Lahham

TL;DR
MELK is a kinase overexpressed in many cancers, linked to aggressive behavior and treatment resistance, but its role as a universal driver is debated.
Contribution
The paper reviews MELK's biological functions, therapeutic potential, and controversies, proposing it as a conditional oncogenic vulnerability.
Findings
MELK overexpression correlates with poor prognosis and aggressive cancer traits.
MELK inhibitors show promise but have limitations and context-specific effects.
MELK may be a conditional vulnerability in p53-deficient TNBC and glioma stem cells.
Abstract
MELK is a serine/threonine kinase that regulates cell cycle progression, proliferation, apoptosis, migration, and stemness. MELK is overexpressed in many solid and hematologic malignancies. Furthermore, high MELK levels correlate with aggressive disease, poor survival, treatment resistance, and cancer stem-like features. Several small-molecule inhibitors and RNAi-targeting MELK have suppressed the development and progression of various cancers. OTSSP167 has shown promising results in phase I and II clinical trials. However, a study employing CRISPR/Cas9 knockout in breast cancer cell lines reported that loss of MELK did not impair proliferation. The review concludes that MELK is a robust prognostic biomarker of proliferation and aggressive behavior across multiple cancers but is not a universal driver of tumor growth. Discrepancies may arise from off-target effects of RNAi or…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
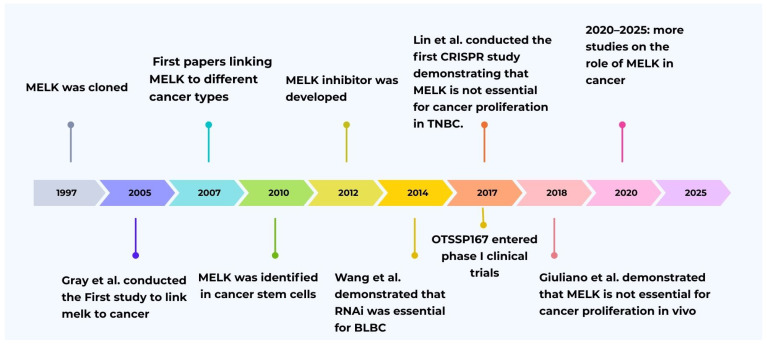 Figure 1
Figure 1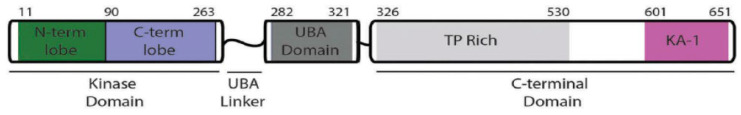 Figure 2
Figure 2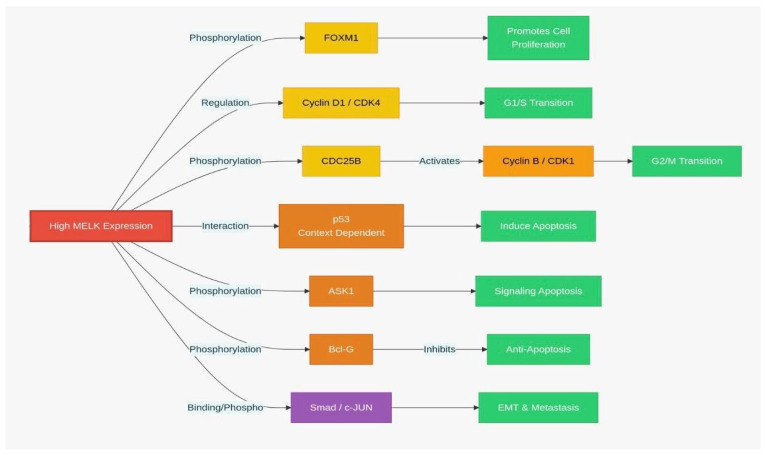 Figure 3
Figure 3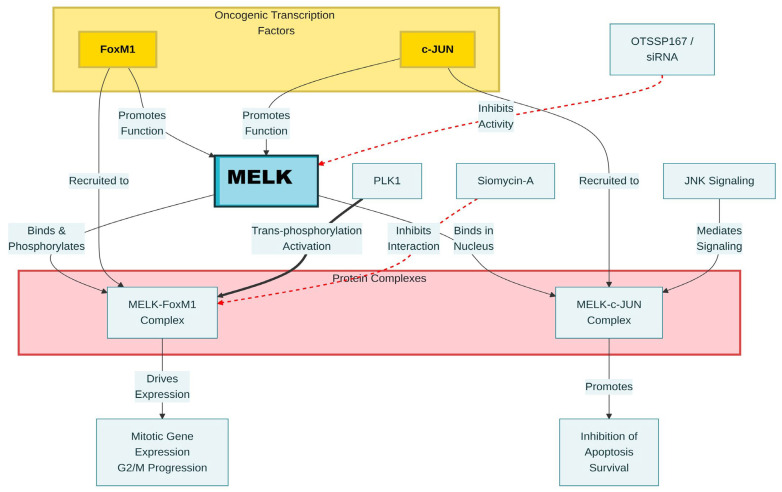 Figure 4
Figure 4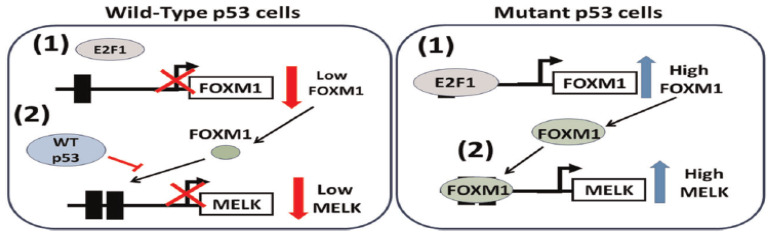 Figure 5
Figure 5| Cancer Type | Prognostic Value | Signaling Pathways | Down Regulation | Key Preclinical Findings | |
|---|---|---|---|---|---|
| Breast cancer (especially TNBC) | Upregulated; highest in aggressive TNBC subtype [ | Worse OS. | G2/M arrest [ | Knockdown/OTSSP167 | |
| Glioma | Overexpressed [ | More aggressive behavior. | G2/M arrest [ | Suppresses {GSC s} more effectively. Enhances radiotherapy response [ | |
| Lung cancer (NSCLC, LUAD, SCLC) | Overexpressed especially in LUAD [ | Worse prognosis in LUAD. | PLK-CDC-CDK1 | G2/M arrest [ | Inhibition disrupts the cell cycle. |
| Gastric cancer | Frequently Overexpressed in primary GCs [ | Worse Clinical Outcomes: Associated with lymph node involvement, distant metastasis, depth of invasion [ | CSF-1/JAK2/STAT3 | G2/M arrest [ | Knockdown/OTS167 increases chemosensitivity, decreases proliferation [ |
| Colorectal cancer | Overexpressed; correlated with tumor stages [ | Worse Prognosis: invasion, metastasis, radiotherapy, chemotherapy resistance [ | AKT-FAK/Src Pathway | G2/M arrest [ | Knockdown reduces proliferation, migration, and invasion [ |
| Melanoma | Overexpression [ | Worse prognosis, invasion, metastasis [ | MAPK pathway | G2/M arrest [ | Knockdown/OTS167/ |
| Hepatocellular carcinoma | Overexpression [ | Worse OS and DFS. Correlate with advanced stage and grade [ | G2/M arrest [ | Knockdown | |
| Bladder cancer | Overexpression [ | Worse prognosis, invasion, and metastasis [ | ATM/CHK2/p53 signaling [ | G1/S arrest [ | Knockdown/OTS167 induces G1/S cell cycle arrest [ |
| Kidney cancer | Overexpression [ | Worse prognosis, proliferation, and metastasis [ | PRAS40/mTORC1 | G2/M arrest [ | Knockdown/OTS167/OTS514 reduces proliferation, enhances apoptosis [ |
| Cervical cancer | Overexpression [ | Worse prognosis, proliferation, Th1/Th2 imbalance [ | PD-L1 | G2/M arrest [ | |
| Ovarian cancer | Overexpression [ | Worse prognosis, tumor aggressiveness, progression, decreased survival [ | G2/M arrest [ | OTSSP167 inhibits growth, increased the sensitivity to carboplatin [ | |
| Endometrial cancer | Overexpression in advanced stages [ | Worse Prognosis Marker of Aggressive [ | JAK2/STAT3 | G2/M arrest [ | Knockdown/OTSSP167 inhibits growth, |
| Prostate cancer | Upregulated in high-grade PC [ | Worse prognosis enhances the survival of PC cells [ | TOP2A, AURKB, UBE2C, | G2/M arrest [ | OTSSP167 induce apoptosis [ |
| Osteosarcoma | Upregulated [ | Worse prognosis; enhances cell survival, proliferation, and metastasis [ | PI3K/Akt/mTOR signaling pathway regulates PCNA and | G2 and S phases [ | Knockdown/OTSSP167 reduced proliferation, migration, and invasion. |
| Chronic lymphocytic leukemia (CLL) | Overexpression [ | Worse prognosis | G2/M arrest [ | Knockdown/Knockout/OTSSP167 | |
| Acute myeloid leukemia | Overexpression [ | Worse prognosis | G2/M arrest [ | Knockdown/OTSSP167 | |
| Multiple myeloma | Overexpression [ | Worse prognosis | G2/M arrest [ | Knock-down/OTSSP167 reduced myeloma cell proliferation and survival [ | |
| Diffuse large B-cell lymphoma and mantle cell lymphoma | Overexpression [ | Worse survival in R-CHOP-treated patients [ | CDC25B activates CDK1 | G2/M arrest [ | Knock-down/OTSSP167 |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsFOXO transcription factor regulation · Endoplasmic Reticulum Stress and Disease · Cancer Mechanisms and Therapy
1. Introduction
MELK gene is a member of the Snf1/AMPK serine/threonine kinase family. MELK cDNA was initially cloned in 1997 (Figure 1) and was initially recognized through its expression among several early embryonic cellular stages [1,2]. This initial finding implicated MELK in essential developmental processes, including embryogenesis and cell cycle regulation [3]. Additionally, it serves as a multifunctional kinase, participating in various biological and cellular processes. These functions include the stringent regulation of cell cycle progression [4], enhancement of cell proliferation [3], enhancement of mitosis [5], modulation of apoptosis [3,6], promotion of cell migration [7], and support for cell renewal [4]. Furthermore, MELK plays a role in cancer cells’ survival, growth, invasion, metastasis, and the regulation of the tumor microenvironment [5]. The extensive body of research has reported elevated MELK levels across various cancer types, with MELK overexpression frequently correlating with unfavorable prognostic outcomes and promoting tumorigenesis by inhibiting apoptosis [6,7]. Therefore, one can envision that MELK knockdown or inhibition may induce apoptosis and disrupt cancer cell proliferation [8]. Indeed, it has been shown that the knockdown of MELK enhances the sensitivity of cancer cells to radiotherapy [8,9]. However, few studies have opposed the findings regarding the MELK gene’s involvement in cancer [10,11,12]. Therefore, we aim to provide a comprehensive overview of MELK’s role in key functions, including proliferation, metastasis, apoptosis, and oncogenesis, as well as the role and therapeutic implications of MELK inhibitors. Finally, the contrasting viewpoints concerning MELK’s involvement in cancer proliferation and the potential of MELK inhibitors to impede cancer cell growth.
2. Structure of MELK
MELK gene is located on chromosome 9p13.2. The gene sequence comprises 2501 base pairs and includes 22 exons, encoding 651 amino acids that result in a 70 kDa protein [1,2]. MELK is very similar across species, from humans to C. elegans, and the crystal structure of a human MELK fragment comprises three distinct domains (Figure 2). The N-terminal Ser/Thr kinase domain, ubiquitin-associated (UBA) domain, and C-terminal regulatory region [1,18]. The N-terminal Ser/Thr kinase domain is a catalytic domain with kinase activity that phosphorylates target proteins at their serine or threonine residues [1,19]. The C-terminal domain serves as a regulatory domain, and the structure consists of a segment rich in proline and threonine (TP-rich) and a kinase-associated segment (KA1) [1,18]. The C-terminal KA1 domain has been identified as auto-inhibitory, capable of interacting and controlling the N-terminal of the AMPK-related kinases [1]. Furthermore, it contains a membrane-association domain that binds acidic phospholipids, such as phosphatidylserine, thereby facilitating kinase interactions with substrates or signaling complexes located within the membrane [20,21]. The TP-rich region consists of several phosphorylation sites, with specific Thr phosphorylation in this region being essential for MELK’s inhibition of spliceosome assembly. Additionally, the TP-rich domain is phosphorylated in mitotically arrested cells and facilitates binding to the FHA (forkhead-associated) domain of the transcription and splicing factor nuclear inhibitor of protein phosphatase 1 (NIPP1) [1,22]. The Ubiquitin-Associated (UBA) domain of MELK is a key regulatory part that is positioned right next to the N-terminal Ser/Thr kinase domain. It is well established that classical UBA domains bind ubiquitin; however, the UBA domain in MELK (and other AMPK-related kinases) lacks this property and binds ubiquitin poorly. Instead, its main function is to control the kinase domain’s catalytic activity and make sure it is in the right conformational shape [18,23]. MELK is regulated by the UBA domain, which functions as an obligatory structural cofactor. It binds to the back of the kinase N-lobe, stabilizing the kinase C-helix in the active conformation and thereby enabling catalytic activity [20,24]. Additionally, it is shown that eliminating the UBA domain from MELK makes the enzyme inactive. This indicates that the UBA domain is not just a regulatory element; it is also essential for MELK’s catalytic activity to be expressed [1].
3. Role of MELK in the Cell Cycle and Apoptosis
MELK is expressed in several tissue types via distinct mechanisms [26]. MELK expression is observed mainly in proliferative cells like embryonic cells, spermatogonia, and oocytes [13,27]. During these stages, MELK expression is tightly regulated, indicating its critical role in determining cell fate and differentiation [28]. MELK expression is significantly associated with the cell cycle. Throughout the cell cycle, MELK mRNA and protein levels fluctuate. Levels peak during the G2/M phases and subsequently decrease after cell division [9]. Its expression is low in non-growing organs and high in dividing tissues, and it is also linked to well-known proliferation markers, such as MKI67 (a nuclear protein widely used as an indicator of cell proliferation). MELK expression is strongly associated with mitotic activity in human cancers [12].
MELK also promote the G1/S transition in the cell cycle [29]. Overexpression of MELK has been shown to increase cyclin D1 and CDK4 levels, which are essential for cell transition from G1 to S phase and for initiation of DNA replication (Figure 3) [29,30]. Conversely, inhibiting MELK by OTSSP167 prevents cell division and transitions from G1 to S phase by decreasing the levels of cyclin E1 and cyclin D1 (25). Furthermore, MELK influences the ATM/CHK2/p53 pathway during the G1/S transition. MELK inhibition activates this pathway, leading to cell-cycle arrest at the G1/S phase. Regulation of the MELK can activate p53 and p21 downstream, thus effectively inhibiting cyclin-dependent kinase (CDK) activity and transcription factors (E2Fs), which are essential for DNA replication during the G1/S phase [29]. However, the interaction between MELK and p53 is complicated. MELK has been reported to phosphorylate Ser15 at amino-terminal transactivation domain of p53, thereby inhibiting the G1/S phase transition. This phosphorylation enhances p53 stability and activity, potentially leading to cell cycle arrest and apoptosis [5]. This apparent contradiction suggests that MELK’s role in regulating p53 is context-dependent. In certain cancer situations or when cells are under significant stress, MELK may activate p53 to induce apoptosis or halt the cell cycle as a protective mechanism. However, in other cases, such as when MELK is overexpressed, which frequently occurs in various malignancies, its primary function may shift towards promoting growth and facilitating the G1/S transition. This shift can occur by bypassing p53-mediated checkpoints or by engaging alternative pathways that counteract the effects of p53 [3,5,31]. This illustrates the complexity of MELK’s signaling network; the effect of this factor on promoting or inhibiting is an ongoing area of research, with evidence supporting both functions. This demands further investigation into its role in apoptosis.
Additionally, MELK is pivotal in activating Fork head Box M1 (FOXM1), a critical transcriptional and carcinogenic factor that controls the expression of numerous mitotic transcription factors [3,32]. Expression of FOXM1 is upregulated in various cancer types, including breast, liver, prostate, brain, lung, colon, and pancreatic cancers [32,33]. The activation of the MELK-FOXM1 axis increases the transcription of Enhancer of zeste homolog 2 (EZH2), which in turn inhibits the transcription of differentiation genes, thereby promoting cancer cells in preserving their stem-like properties [34]. Additionally, MELK interacts with other critical transcription factors, including c-JUN [26], Early region 2 binding factor (E2F1) [35], protein tyrosine phosphatase (CDC25B) [36], zinc finger protein analog (ZPR9), nuclear inhibitor of serine/threonine protein phosphatase-1 (NIPP1), transforming growth factor-β (TGF-β), signal transduction Smad protein, apoptosis signal-regulated kinase 1 (ASK1), and pro-apoptotic gene Bcl-G (Figure 3) [37].
4. Evidence Supporting the Role of MELK in Cancer
The initial investigation regarding MELK expression in tumor tissue and its connection to tumor growth was conducted by Daniel et al. in 2005 [5]. High MELK expression has been studied in many types of cancer, such as colon, breast, ovary, and lung cancer, compared to normal tissue [5]. This study was followed by numerous other studies over the next two decades, which demonstrated high MELK expression in many cancer types, including glioblastoma, breast cancer, prostate cancer, gastric cancer, liver cancer, ovarian cancer, and endometrial cancer (Figure 1) [5,14]. Conversely, MELK expression knockdown by siRNA has been shown to inhibit proliferation in cervical, breast, colorectal, pancreatic cancer cells, and other cancer types [13]. This led to the development of MELK inhibitors; in 2012, OTSS167 was found to effectively inhibit growth and induce apoptosis across multiple cancer types, including breast cancer, acute myeloid leukemia, and small-cell lung cancer (Figure 1) [15,38]. Moreover, high MELK expression is associated with aggressive disease and poor clinical outcomes for different cancer types and correlatively decreased overall survival (OS), recurrence-free survival (RFS), and distant metastasis-free survival (DMFS) (Table 1) [39]. However, the mechanisms by which MELK facilitates aggressive tumor growth remain incompletely elucidated [40].
Regarding molecular mechanisms, in cancer stem cells, MELK function is promoted by other oncogenic transcription factors, such as FoxM1 and c-JUN [16,26]. MELK interacts with FoxM1 in glioma stem-like cells (GSCs), promoting FOXM1 phosphorylation and activation, thereby increasing the expression of mitotic regulatory genes in GSCs. Furthermore, the activation of FOXM1 driven by MELK is regulated through the binding and subsequent trans-phosphorylation of FOXM1 by the kinase PLK1 [16]. MELK interacts with c-JUN in the nucleus, forming a GSC-specific protein complex that is not observed in normal cells. JNK-mediated MELK/c-JUN signaling inhibited apoptosis and enhanced the survival of GSCs. Inhibiting MELK-FOXM1 interaction by treatment with siomycin-A leads to cell cycle arrest in G2/M phase [16]. Furthermore, the GSCs’ sensitivity to radiotherapy can be improved via MELK knockdown using siRNA or shRNA, leading to apoptosis of GSCs and a reduction in tumor formation, or treatment with the MELK inhibitor (OTSSP167), thereby enhancing their therapeutic response (Figure 4) [4,26,41].
In the same context, MELK, regulated by E2F1 and FOXM1 in breast cancer, is contingent upon the mutational status of p53. This mechanism has been explicitly defined, especially in p53-mutant triple-negative breast cancers (TNBCs). E2F1 shows decreased binding to the FOXM1 promoter in wild-type p53 breast cancer cell lines, thereby lowering FOXM1 expression and, consequently, reducing MELK expression. Conversely, the absence of wild-type p53 activity results in increased MELK levels in p53-mutant breast tumors. (Figure 5) [42]. Nevertheless, Mutated p53 failed to inhibit FOXM1 and MELK expression, thereby promoting tumor development in TNBCs. This suggests that mutant p53 is responsible for inducing MELK expression, which coincides with the growth of TNBC [9]. Moreover, MELK knockdown triggers G2 cell-cycle arrest by reducing cyclin B1 expression, increasing p27 expression, and JNK phosphorylation in TNBC cell lines. Additionally, in non-TNBC cell lines, it induces G1 cell-cycle arrest by decreasing the expression of p21, cyclin B1, and cyclin D1, and by reducing cdc2 phosphorylation. This suggests that the tumorigenesis mechanism of MELK in TNBC differs from that in non-TNBC cells. Furthermore, MELK knockdown via RNAi preferentially reduced BLBC cell proliferation and induced apoptosis, resulting in smaller cell size and increased radiation sensitivity in TNBC [9,43]. Additionally, MELK inhibitors such as OTSSP167 downregulate FOXM1 expression and its target gene. (Figure 4) [23].
In bladder cancer (BC), MELK is markedly overexpressed in BCa cells obtained from patient tissues compared with normal tissues. MELK expression continues to increase in tandem with tumor progression and is associated with cell proliferation and migration. BC exhibits a unique characteristic compared with other types. MELK suppression leads to cell cycle arrest at the G1/S phase by interacting with the ATM/CHK2/p53 pathway. MELK downregulation led to sequential phosphorylation of ATM and CHK2, as well as upregulation of p53 and p21 [29].
In chronic lymphocytic leukemia (CLL), MELK is overexpressed in CLL cells and in cell lines (EHEB and MEC1) relative to normal B cells. This overexpression contributes to increased CLL cell survival by decreasing apoptosis and promoting G2/M phase transition [44,45]. In CLL, the findings differed from those of previous studies about the effect of CRISPR/Cas9 on MELK. In addition to knockdown via siRNA, MELK Knockout mediated by CRISPR/Cas9 enhances apoptosis and suppresses proliferation in CLL cells [45]. A substantial body of research indicates that the overexpression of MELK in many cancer types is associated with poor prognosis, cancer cell proliferation, metastasis, and chemo-radiotherapy resistance. Furthermore, MELK depletion via RNA interference exhibits antiproliferative effects.
5. MELK Inhibitors
Chemotherapy has served as the most fundamental and prevalent treatment approach in the fight against cancer in recent decades [5,7]. Recent studies have focused on the MELK gene inhibitors using small molecules, such as OTS167 [18], Cyclosporine A [74], MELK 8a, MELK 8b [75], HCTMPPK [74], MELK-T1 [23], and Phillygenin (PHI) [75].
6. OTS167 (OTSSP167)
OTSSP167 also known as OTS167 is composed of a “((1,5-naphthyridine core that contains methylketone at the 3-position, trans-4-((dimethylamino)methyl)) cyclohexylamino at the 4-position, and 3,5-dichloro-4-hydroxyphenyl at the 6-position))” [76]. OTSSP167, with an IC50 of 0.41 nM, is a novel inhibitor that specifically targets MELK, efficiently suppressing the development and progression of various types of cancer. OTSSP167 has shown promising results in phase I and II clinical trials in AML and breast cancer, making it the only MELK inhibitor presently available [37,45]. Additionally, OTSSP167 has the effect of reducing the expression of FOXM1, cyclin B1, and CDK1, causing cessation of cell cycle progression and arrest at the G2/M phase [44]. On the other hand, OTSSP167 treatment led to cell cycle arrest at the G2/M phase by upregulation of p21 expression [33]. Alternatively, new evidence suggests that OTSSP167 can inhibit cell proliferation via a non-target-specific mechanism since the tumor was still responsive to the compound even after MELK knockout [11]. The ambiguity surrounding the primary therapeutic target of a potent compound poses a significant challenge in drug development, despite its advancement in clinical trials.
Accumulated research conducted over the last decade has demonstrated the impact of OTSSP167 on inhibiting the proliferation of several types of tumor cell lines by targeting MELK in a dose-dependent manner, including NSCLC, SCLC [15,49], TNBC [9], colorectal cancer [77], kidney cancer [60], cervical cancer [78], ovarian cancer [65,79], prostate cancer [15], osteosarcoma [69], CLL [45], AML [38], and Multiple myeloma [71]. Additionally, OTS167 demonstrated a substantial in vivo inhibitory effect on tumor growth across various human cancer xenograft mouse models [49], the effect demonstrated in prostate cancer [67], endometrial carcinoma [57], neuroblastoma [17] TNBC, lung cancer, prostate cancer, pancreatic cancer [15], adrenocortical cancer [80], and gastric cancer [50].
Furthermore, OTSSP167 has been combined with other compounds to enhance the inhibitory effect on MELK. The combination of OTSSP167 with bortezomib results in more potent inhibition of the proliferation of breast cancer cells [81]. Likewise, the OTSSP167 and cyclin-dependent inhibitor (RGB-286638) combination was used in adrenocortical cancer treatment. This synergism yielded a markedly greater antiproliferative impact, increased caspase-dependent apoptosis, and downregulated MELK expression [80]. The research suggests that OTSSP167 is a novel and effective compound, warranting further in vivo testing for cancer treatment and clinical trials.
7. Cyclosporine A
Due to its high efficacy, the immunosuppressant medicine cyclosporine A (CsA) has found widespread application in organ transplantation [82]. In addition, CsA demonstrates antineoplastic effects against various cancer types, including prostate cancer [83,84]. In prostate cancer, a critical transcription factor E2F8 promotes cancerous characteristics and influences poor prognosis. Furthermore, the MELK-E2F8 signaling axis is integral to the biology of prostate cancer. Additionally, CsA significantly reduced E2F8 expression. Therefore, in cases of prostate cancer, E2F8 is a potential target for therapeutic intervention, and MELK is a crucial component in regulating the expression of E2F8. Thus, inhibiting either E2F8 or MELK improves the sensitivity of prostate cancer cells to androgen receptor-blocking treatment [83].
8. Tetramethyl Pyrazine Chalcone Hybrid-HCTMPPK
Tetramethyl pyrazine (TMP), often known as ligustrazine, is a potent alkaloid monomer with very effective bioactive components. Extensive studies have conclusively shown that TMP and its derivatives have significantly inhibited the growth of various cancer cells, including those in liver cancer [85,86], lung cancer [87], colorectal cancer [88], and breast cancer [89]. HCTMPPK (a derivative of TMP) was subjected to molecular docking with the MELK, AURKA, and JUN proteins. The interaction between HCTMPPK and MELK genes significantly reduced MELK expression and hindered the progression of NSCLC. Although HCTMPPK has been proven to inhibit MELK expression, the exact mechanisms involved remain unclear [74]. These studies indicate that HCTMPPK may act as an effective anti-tumor chemotherapeutic agent.
9. MELK 8a, 8b
The compound MELK 8a, 8b is alternatively referred to as “((1-methyl-4-[4-[4-[3-(piperidin-4 ylmethoxy) pyridin-4-yl] pyrazol-1-yl] phenyl] piperazine))”, also known as Novartis MELK inhibitor 8a (NVS-MELK8a) and (NVS-MELK8b). The compounds inhibit cell proliferation and cell cycle in MELK-dependent MDA-MB-468 cells [90]. Furthermore, MELK 8a demonstrates high selectivity as a MELK inhibitor, representing the first dependable alternative to OTSSP167 for functional studies of MELK. Additionally, MELK 8a reduces the viability of and diminishes the growth of TNBC cell lines [91]. In addition, it has high selectivity as a MELK inhibitor; this inhibition results in a postponement of the mitotic entrance, which is consistent with a transient G2 arrest state [91]. Furthermore, MELK 8a decreased SQSTM1 phosphorylation and inhibited the NF-κB pathway, consequently suppressing the proliferation of melanoma cells [5,35]. These studies indicate that MELK 8a and 8b inhibit the proliferation of cancer cells with high MELK expression; therefore, additional research is necessary to clarify the mechanisms of this inhibition.
10. MELK-T1
The compound MELK-T1 is alternatively referred to as “2-methoxy-4-(1H-pyrazol-4-yl)-N-(2,3,4,5-tetrahydro-1H-3-benzazepin-7-yl) benzamide))”. MELK-T1 demonstrates significant and selective inhibition of the MELK domain, rapidly decreasing endogenous MELK levels by causing it to break down through a process that depends on the proteasome, and inhibiting the growth of MCF-7, a breast adenocarcinoma cell line. On the other hand, MELK-T1 is classified as a type I ATP-mimetic inhibitor, binds to MELK, and activates the degradation of the MELK protein by stabilizing the ATP-bound conformation [23]. Furthermore, MELK-T1 affects various mechanisms, significantly inducing p53 phosphorylation, prolonging the upregulation of p21, and downregulating FOXM1 and its target genes. This is consistent with the previously reported effects of OTSSP167 on AML cell lines [15,23]. The inhibition and depletion of MELK protein through MELK-T1 treatment could allow cancer cells to recognize and respond to DNA damage once again, thereby enhancing tumor sensitivity to radiotherapy and chemotherapy [23]. Ultimately, the dual mechanism of inhibiting enzyme function while reducing its cellular abundance may yield a more effective and lasting therapeutic outcome compared to inhibitors that exclusively target catalytic activity.
11. The Controversy: The Crucial Role of MELK in Cancer Growth (RNAi vs. CRISPR/Cas9 Methods)
RNA interference (RNAi) and Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)/CRISPR-associated protein 9 (Cas9) systems are two innovative gene-silencing technologies that have transformed molecular biology research and therapeutic advancement. While both can precisely regulate gene expression, they differ fundamentally in their methods and advantages for specific applications. RNAi is a mechanism for transient, potentially reversible gene knockdown at the post-transcriptional level via mRNA degradation. CRISPR Cas9, on the other hand, is an appropriate technology for more permanent knockouts that involve gene editing directly within DNA [92].
The use of RNAi and small molecule inhibitors demonstrated MELK’s crucial role as a vital therapeutic target due to the issue of overexpression in various cancers and its association with severe disease phenotypes, chemotherapy resistance, cancer stem cell renewal, and overall cancer proliferation, resulting in the progression of MELK inhibitors to clinical trials. Nonetheless, the definitive role of MELK in cancer cell proliferation remains a topic of significant debate within the scientific community, characterized by inconsistent results from various experimental methodologies. The variance can be attributed to the researchers’ utilization of a CRISPR/Cas9 methodology, which completely knocked out MELK.
However, few research studies have emerged that contradict the findings of prior investigations. Wang Y. et al. (2014) [9], concluded that the proliferation of BLBC cells requires MELK activity, whereas luminal breast cancer cell proliferation does not. In addition, post-MELK knockdown via RNAi has shown that Exogenous WT MELK expression can rescue the growth effects in both cells and tumors [9,48]. Later, Lin A. et al. (2017) [11] used a different technology, CRISPR/Cas9, to generate frameshift mutations in the MELK gene, aiming to investigate the impact of MELK knockout on cancerous cell lines (Figure 1). Pooled MELK-null, seven triple-negative breast cancer cell lines, were created using gRNAs. These lines were compared with the negative control Rosa26 deletion control cells, and the proliferative activity of MELK cell lines, whether they were wild-type or mutant, remained unchanged. Based on these findings, MELK expression is not essential for TNBC, and it does promote cancer cell proliferation [11]. These findings prompt us to reconsider whether MELK inhibitor OTS167 can prevent cancer cell proliferation. If the MELK inhibitors stop tumor cell proliferation and tumor cells are not MELK-dependent, then OTS167 is successful in inhibiting tumor cell proliferation or its cellular target genes, other than MELK. This may lead us to the conclusion that OTS167’s anti-proliferative actions are not due to its suppression of MELK [11]. In a different study in 2017, Huang H et al. [10] used integrated chemical and genetic modifications. Researchers have employed a novel method for protein degradation that utilizes chemical agents, CRISPR, and RNAi to investigate MELK as a potential therapeutic target for BBC therapy. Also, slight molecule inhibition, gene deletion, or MELK exhaustion under standard culture conditions did not significantly affect cellular proliferation. Both gene editing and pharmaceutical suppression of MELK in breast cancer cell lines failed to inhibit cell proliferation in vitro [10]. The study’s discrepancy with previous research prompted researchers to investigate the selection effects of OTSSP167 and the potential off-target consequences of short hairpins targeting MELK. Regarding this issue, researchers compared the sensitivity to selective MELK inhibitors and OTSSP167 in wild-type (WT) and MELK knockout (MELK−/−) cells. Their conclusion indicated that after treatment with OTSSP167, there are no differences in cell viability, demonstrating that the effect of OTSSP167 on the viability of MDAMB-468 cells is not attributable to MELK inhibition and that its target is different from MELK. The findings indicate that under typical culture conditions, suppressing or depleting MELK alone has no adverse effect on BBC cell line proliferation [10]. In a 2018 study, Giuliano et al. observed that the overexpression of MELK alone was insufficient to induce transformation in immortalized cell lines. Despite findings from CRISPR/Cas9 applications, Zhang et al. reported contradictory results in a 2018 study. MELK knockout achieved by CRISPR/Cas9, along with siRNA knockdown, enhances apoptosis and suppresses proliferation in CLL cells [45]. The findings indicate that MELK is not essential for cancer cell proliferation, both in vivo and in vitro, and indicate that the immediate absence of MELK does not result in any notable impairment of cell viability, proliferation, or drug resistance. Furthermore, the results show that MELK knockout cells have not developed mutations that make cells more tolerant to the lack of MELK [12,93]. Research utilizing CRISPR technology indicates that MELK does not play a significant role in the proliferation of cancer cells and is not a viable therapeutic target. These findings, which show no proliferation after MELK knockout with CRISPR, do not conclusively support these claims. Even though these studies have conclusively shown that MELK is not essential for the proliferation of cancer cells, no actions have been taken by them to argue against the findings that MELK knockdown by RNAi can be rescued by exogenous WT MELK expression, indicating that MELK is essential for proliferation and that the therapeutic potential of decreasing MELK should not be disregarded at this time, especially when used in combination with other chemotherapeutic medications [14].
12. Discussion
The broad literature analyzed in this paper has shown that MELK, a multifaceted oncogenic kinase, plays a role in cancer biology that has not been fully comprehended over decades of study. The main paradox that the overall analysis elicits is the drastic opposition between the functional research in utilizing RNAi and the small-molecule suppressors on the one hand and the genetic deletion through CRISPR/Cas9 on the other. This contradiction essentially questions our perception of MELK as a target of therapy and is an aspect that needs to be interpreted cautiously.
13. The Experimental Methodology Paradox
The potential mismatch between knockdown experiments using RNAi and the CRISPR/Cas9 knockout is the most obvious gap in the research of MELK. Initial studies by Wang et al. 2014 had convincingly proved that MELK activity was required to sustain the growth of BLBC, and exogenous wild-type MELK was able to rescue growth-inhibitory phenotypes caused by RNAi [9]. Later CRISPR/Cas9 studies by Lin et al. 2017, Huang et al. 2017 failed to confirm these results, finding that the complete elimination of MELK by frameshift mutagenesis in TNBC cell lines had little or no effect on cell proliferation [10,11]. This methodological distinction is not trivial. RNAi approaches generate temporary, partial knockdown with residual MELK expression, whereas CRISPR/Cas9 induces complete, permanent knockdown. It is possible to interpret such divergent results in three non-mutually exclusive mechanisms: (1) off-target effects of RNAi hairpins that lead to spurious phenotypes; (2) off-target inhibition of other kinases by MELK inhibitors that include OTSSP167; (3) cell line-dependent dependencies or adaptive responses that are not compensated for by MELK loss through redundant signaling pathways [14].
There is strong evidence on the off-target hypothesis. Huang et al. have shown that when MELK-knockout cells were treated with OTSSP167, the same growth-inhibitory effects were observed as when using wild-type cells, a fact that powerfully suggests the anti-cancer effect of the compound, which appears to be present in other targets besides MELK [10]. This observation leads to a radical re-evaluation of whether the reported anti-tumor effects of MELK inhibition are due to MELK-specific actions or to the polypharmacology of effectors targeting a range of kinases. The clinical implications are high because the progression to the Phase I/II clinical trials of OTSSP167 was based on the premise of an on-target mechanism of action.
14. Context-Dependent MELK Functions
The literature indicates that MELK has a context-dependent role in cancer proliferation and that the status of p53 (mutation or not), and the type of molecular subtype of cancer, significantly determine the role of MELK in cancer proliferation. Such as in TNBC, MELK overexpression is related to mutant p53, compared with wild-type p53 in luminal breast cancers [9]. The mechanistic basis of this regulatory difference involves E2F1 and FOXM1, whose binding dynamics at the MELK promoter differ between p53-wild-type and p53-mutant cells. Mutant p53 does not suppress FOXM1 expression, leading to elevated MELK levels that stimulate TNBC tumor growth. On the other hand, in breast cancer with wild-type p53, E2F1-p53-based FOXM1 suppressions cause decreased EMLK [9,42]. This finding indicates that MELK can more likely operate as a conditional vulnerability—a treatment target that is not universally active on all cancers but is most likely to be relevant in most instances of p53-deficiency, where the MELK-controlled proliferation pathways are the most common.
Furthermore, the observation that MELK knockdown produces differential effects across cancer subtypes further supports this hypothesis. In TNBC and glioma stem cells, MELK depletion triggers G2/M phase arrest and apoptosis, whereas in bladder cancer and non-TNBC cell lines, G1/S phase arrest predominates [9,29,43]. These mechanistic differences indicate that MELK interacts with different regulatory networks depending on the cellular context. MELK in TNBC interacts with the MELK-FOXM1 axis that regulates mitotic transcription factors and fosters stem-like characteristics in a coordinated manner using EZH2 to suppress differentiation genes [9,43]. In GSCs, MELK interacts with c-JUN to form a GSC-specific protein complex that enhances JNK-induced signaling and inhibits apoptosis [29]. The specificity of these interactions suggests that MELK is not a universal oncogene; instead, it functions as a pathway node that becomes critical in specific malignant contexts in which upstream oncogenic signals converge on MELK-dependent effectors.
15. MELK as a Prognostic Biomarker
Despite remaining contentious that MELK is necessary in cancer growth, the increasing body of evidence in several cancer types has shown that there is a strong and consistent relationship between MELK overexpression and poor clinical prognosis. There is an association of high MELK expression with poor overall survival rates, low recurrence-free survival rates, and high distant metastasis-free survival rates in breast cancer and glioma, as well as lung cancer, gastric cancer, colorectal cancer, ovarian cancer, endometrial cancer, kidney cancer, prostate cancer, and hematologic cancer (Table 1). Such a conclusion made by finding a strong correlation in multiple types of cancer shows that MELK is a powerful predictor of malignancy and indicative of proliferation potential. Nonetheless, tumor growth may not be required in every situation. The robust correlation between MELK expression and the proliferation marker MKI67 in human tumors indicates that MELK is an indirect marker of mitotic activity rather than a specific, context-dependent marker of MELK activity [12]. Additionally, it appears that MELK prognostic value is high, especially in aggressive forms, including TNBC, high-grade glioma, and SCLC, where MELK overexpression is linked with poor prognosis and predicts higher aggressiveness. The clinical implications of such a difference are that a biomarker that has a high probability of predicting aggressive disease and poor prognosis has a high clinical value in risk stratification, treatment planning, and patient monitoring, whether it is considered as a direct therapeutic activity or not.
16. Therapeutic Implications and Combination Strategies
The literature on the MELK inhibitor, mainly focusing on the research of OTSSP167, demonstrates the potential and challenges in the development of the MELK-targeted therapy. Pre-clinical experiments have always able to prove that OTSSP167 inhibits tumor cell proliferation, increases vulnerability to chemotherapy and radiotherapy and prevents metastatic capacity in a variety of xenograft models [49]. These results justify further studies of MELK inhibition in clinical settings, especially when used as a part of combination therapy. However, the evidence for off-target mechanisms underlying OTSSP167’s anti-tumor activity necessitates a paradigm shift in how these compounds are evaluated.
17. Conclusions
Maternal Embryonic Leucine Zipper Kinase (MELK) is also considered as an important oncogenic protein, and it is overexpressed in most human malignancies. MELK overexpression can be associated with poor prognosis, treatment aggressive manifestation, treatment re-resistance, and stem-like morphologies of tumors. The correlations reached are evidence that MELK is a clinical biomarker of tumor proliferation and malignancy. Despite these observations, the fundamental contradiction between RNAi-mediated studies demonstrating MELK essentiality and CRISPR/Cas9 knockout studies showing MELK independence cannot be reconciled without acknowledging the probability of substantial off-target effects in both experimental methodologies and therapeutic inhibitors. This unresolved paradox mandates a more nuanced interpretation of MELK’s role in cancer biology and necessitates reformulation of therapeutic strategies. Moreover, the anti-tumor activity of OTSSP167 appears to involve off-target mechanisms, further complicating the interpretation of MELK’s role as a direct therapeutic driver.
The following conclusions are supported by the evidence provided in the present paper: First, MELK is an effective and clinically relevant prognostic biomarker of tumor aggressiveness, proliferative ability and poor clinical outcome across many different cancer types. Second, MELK is a conditional oncogenic vulnerability that is most pertinent in particular molecular contexts, specifically, p53-deficient TNBC and glioma stem cells, rather than an all-encompassing oncogenic driver. Third, the antitumor effects of OTSSP167 and related off-target kinase-inhibitory effects probably contribute significantly to antitumor activity and make it challenging to interpret published preclinical data. Finally, MELK inhibition may retain therapeutic utility when combined with chemotherapy, radiotherapy, immunotherapy, or multikinase inhibitors, despite uncertainty regarding on-target selectivity.
In the future, the MELK field requires several critical research methodologies and improvements in clinical strategies. To determine the on-target mechanisms, first, complete kinase selectivity profiling of all MELK inhibitors should be conducted using biochemical assays and cellular studies in isogenic wild-type and knockout backgrounds. Second, the exploration of the MELK activities in the tumor microenvironment, specifically, influence on macrophage polarization, immune cell infiltration, and regulation of checkpoint pathways, is an untapped prospect in the context of deepening the knowledge of MELK-mediated immunosuppression. Third, mechanistic experiments should determine which molecular subtypes and genetic backgrounds in which MELK is a veritable dependency, using patient-derived xenografts, cellular adaptation, and redundancy conclusion primary tumors, and engineered cell lines. Lastly, MELK-targeted agents should be developed for clinical use to prioritize combination therapy with existing agents rather than demonstrating efficacy in monotherapy, given their emerging non-essential status in most settings.
Although the functionality of MELK as a marker of cancer proliferation remains controversial, its persistent correlation with malignant phenotypes, resistance to treatment, and poor clinical outcomes makes MELK a significant biological aspect of aggressive cancers that require further studies and continued investigation. MELK may not meet the requirements of a classical oncogenic driver—a solitary protein the inhibition of which is enough to inhibit tumor development—but is an informative biomarker and a promising target in therapeutic co-targeting in multimodal treatment approaches that target multiple dependencies of cancer cells. Future studies should be able to differentiate the utility of MELK as a prognostic factor and a therapeutic target by defining context-dependent treatment modalities that may be applied using the biology of MELK, but without overgeneralizing its main and context-specific roles in cancer pathogenesis.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Beullens M. Vancauwenbergh S. Morrice N. Derua R. Ceulemans H. Waelkens E. Bollen M. Substrate Specificity and Activity Regulation of Protein Kinase MELKJ. Biol. Chem.2005280400034001110.1074/jbc.M 50727420016216881 · doi ↗ · pubmed ↗
- 2Jung H. Seong H.-A. Ha H. Murine Protein Serine/Threonine Kinase 38 Activates Apoptosis Signal-regulating Kinase 1 via Thr 838 Phosphorylation J. Biol. Chem.2008283345413455310.1074/jbc.M 80721920018948261 PMC 3259894 · doi ↗ · pubmed ↗
- 3Ganguly R. Mohyeldin A. Thiel J. Kornblum H.I. Beullens M. Nakano I. MELK—A conserved kinase: Functions, signaling, cancer, and controversy Clin. Transl. Med.20154 e 1110.1186/s 40169-014-0045-y PMC 438513325852826 · doi ↗ · pubmed ↗
- 4Liu H. Sun Y. Qi X. Gordon R.E. O’Brien J.A. Yuan H. Zhang J. Wang Z. Zhang M. Song Y. EZH 2 Phosphorylation Promotes Self-Renewal of Glioma Stem-Like Cells Through NF-κB Methylation Front. Oncol.2019964110.3389/fonc.2019.0064131380279 PMC 6652807 · doi ↗ · pubmed ↗
- 5Tang B.-F. Yan R.-C. Wang S.-W. Zeng Z.-C. Du S.-S. Maternal embryonic leucine zipper kinase in tumor cells and tumor microenvironment: An emerging player and promising therapeutic opportunity Cancer Lett.202356021612610.1016/j.canlet.2023.21612636933780 · doi ↗ · pubmed ↗
- 6Maes A. Maes K. Vlummens P. De Raeve H. Devin J. Szablewski V. De Veirman K. Menu E. Moreaux J. Vanderkerken K. Maternal embryonic leucine zipper kinase is a novel target for diffuse large B cell lymphoma and mantle cell lymphoma Blood Cancer J.201998710.1038/s 41408-019-0249-x 31740676 PMC 6861269 · doi ↗ · pubmed ↗
- 7Thangaraj K. Ponnusamy L. Natarajan S.R. Manoharan R. MELK/MPK 38 in cancer: From mechanistic aspects to therapeutic strategies Drug Discov. Today 2020252161217310.1016/j.drudis.2020.09.02933010478 · doi ↗ · pubmed ↗
- 8Choi S. Ku J.-L. Resistance of colorectal cancer cells to radiation and 5-FU is associated with MELK expression Biochem. Biophys. Res. Commun.201141220721310.1016/j.bbrc.2011.07.06021806965 · doi ↗ · pubmed ↗