Genetic Diversity and Population Structure in Two Mangrove Species (Sonneratia alba and Sonneratia caseolaris) Across Coastal Areas of Thailand
Supaporn Khanbo, Chaiwat Naktang, Wasitthee Kongkachana, Chutintorn Yundaeng, Nukoon Jomchai, Nattapol Narong, Tamanai Pravinvongvuthi, Pasin Maprasop, Waratthaya Promchoo, Sithichoke Tangphatsornruang, Wirulda Pootakham

TL;DR
This study examines the genetic diversity and population structure of two mangrove species in Thailand, revealing strong geographic clustering and low genetic diversity.
Contribution
The study provides genome-wide insights into the spatial genetic structure of two Thai mangrove species using RAD-seq data.
Findings
Both Sonneratia alba and Sonneratia caseolaris show low genetic diversity and high genetic differentiation between coastal regions.
Population structure is strongly associated with geography, with distinct genetic clusters in the Andaman Sea and Gulf of Thailand.
Genetic differentiation increases with geographic distance in S. caseolaris but not in S. alba.
Abstract
To assess the genetic diversity and population structure of Sonneratia alba and Sonneratia caseolaris in Thailand, we used restriction site-associated DNA sequencing (RAD-seq) to genotype individuals from natural populations along the Thai coast. Based on SNPs identified from the S. alba and S. caseolaris genome sequences, we found low genetic diversity and high genetic differentiation among 107 S. alba and 131 S. caseolaris individuals. Population structure was strongly associated with geography: individuals formed two main genetic clusters corresponding to the Andaman Sea and Gulf of Thailand coasts, as consistently supported by population structure, principal component and phylogenetic analyses. Sonneratia alba Sm. and Sonneratia caseolaris (L.) Engl. are two ecologically important components of mangrove communities in Thailand. However, their population genetic patterns in Thailand…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
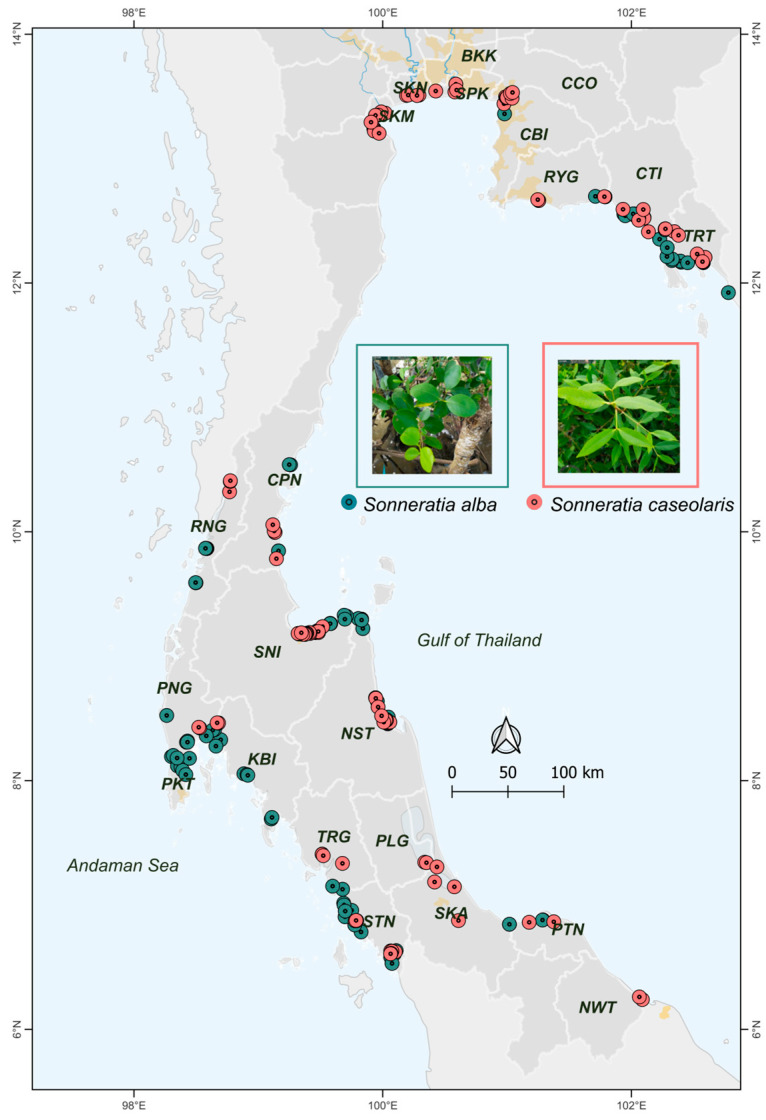 Figure 1
Figure 1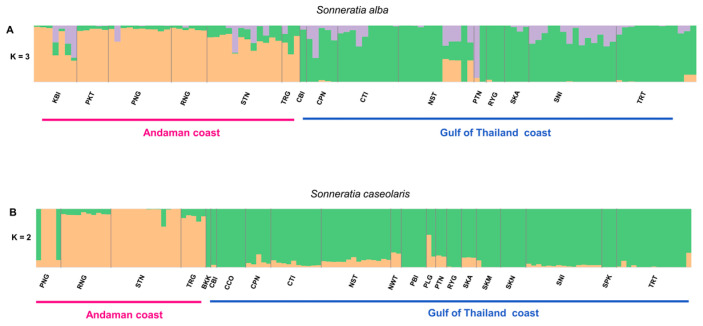 Figure 2
Figure 2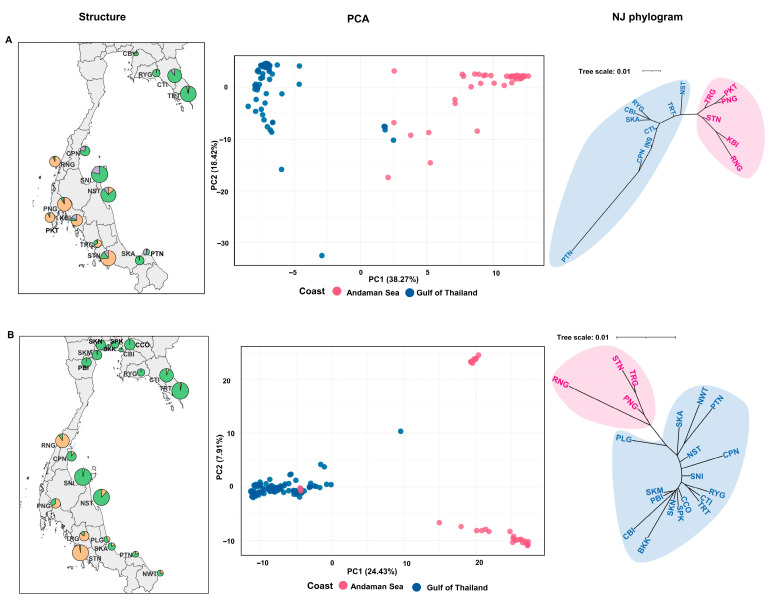 Figure 3
Figure 3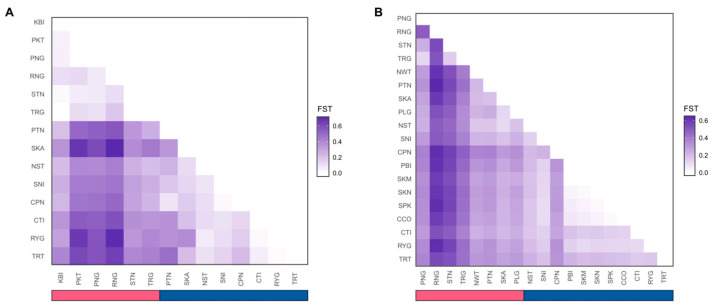 Figure 4
Figure 4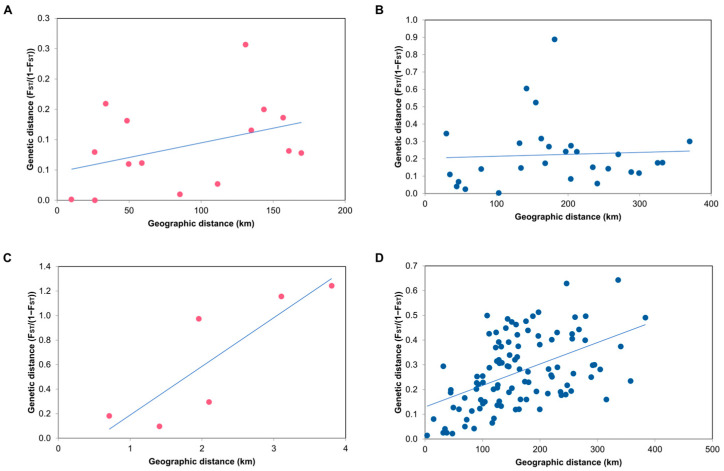 Figure 5
Figure 5- —National Science and Technology Development Agency (NSTDA)
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsCoastal wetland ecosystem dynamics · Marine and coastal plant biology · Genetic diversity and population structure
1. Introduction
Mangrove forests are a key component of coastal ecosystems across tropical and subtropical regions worldwide [1]. They cover approximately 14.7 million hectares and comprise 83 true mangrove species (including 12 hybrids) along with more than 100 associated species [2]. Mangrove forests are among the most important and highly productive coastal ecosystems [3]. They provide numerous ecological functions, including storm surge protection and carbon sequestration for climate change mitigation [4,5]. Mangroves also serve as critical habitats for a wide range of fauna and offer multiple ecosystem services that support coastal communities [6]. In addition, they provide economically valuable resources, such as timber and medicines [7]. Despite their importance, mangroves are vulnerable to various anthropogenic disturbances, including overexploitation, land reclamation, freshwater diversion for irrigation, infrastructure development, and urbanization [8,9]. These activities contribute to habitat loss and erode genetic diversity within mangrove species by reducing effective population sizes, which promotes inbreeding and accelerates diversity loss through genetic drift [10,11,12]. As genetic variation declines, populations may suffer reduced fitness and adaptive potential, increasing extinction risk.
In Thailand, mangrove forests are primarily distributed along the coastlines of the Andaman Sea and the Gulf of Thailand. Mangrove forests are also found on muddy tidal flats at the river mouths [13,14]. Coastal differences in monsoon regimes, currents, and estuarine conditions shape propagule dispersal and the population genetic structure of mangrove populations. According to recent global data on mangrove extent [15], approximately 23% of Thailand’s mangrove forests were lost between 1970 and 2020 along both coastlines [16,17], with the most significant mangrove area losses occurring before 2000. These declines require the implementation of effective strategies for the protection, conservation, and restoration of mangrove ecosystems [18].
The mangrove genus Sonneratia is one of the dominant and ecologically important genera in Thailand [19]. It comprises six species and is widely distributed throughout the Indo-West Pacific region [20]. Sonneratia caseolaris (L.) Engl. is a widespread species, ranging from Sri Lanka through the Malay Peninsula to China and Australia [21,22]. It can naturally grow in both saltwater and freshwater environments [23]. Its closely related species, Sonneratia alba Sm., is distributed from East Africa through Southeast Asia to southern Japan and northeastern Australia [24]. S. alba is highly tolerant to salinity and typically dominates zones along the seaward fringe of mangrove ecosystems [25,26]. In Thailand, S. caseolaris is locally known as Lampoo, while S. alba is referred to as Lampoo Thale [19]. Both species are key structural components of estuarine mangrove forests and provide important ecological and coastal protection services. Although S. alba and S. caseolaris are not currently listed as threatened [12], continued deforestation and degradation of mangrove habitats may lead to long-term genetic risks. Previous studies on the genetic diversity and population structure of S. alba using chloroplast fragments and nuclear genes have shown that this species exhibits low within-population genetic diversity and pronounced genetic structure across the Indo–West Pacific [24]. Wee et al. [27] further characterized the genetic structure and gene flow of S. alba using nuclear microsatellite markers and reported significant east–west genetic differentiation across the Malay Peninsula. In contrast, S. caseolaris populations in Vietnam, evaluated using ISSR markers, exhibit moderate-to-high levels of genetic diversity and cluster into three genetic groups corresponding to their geographic distribution [28]. However, existing knowledge of both species is mainly based on organelle markers or a limited number of nuclear loci. These marker systems provide insufficient resolution for detecting contemporary gene flow, cryptic subdivision, and subtle population divergence that are critical for conservation planning.
In addition to population genetic structure, several studies in mangrove species have reported genetic variation associated with adaptive traits such as salinity tolerance, growth performance, and responses to environmental stress, primarily using candidate gene approaches or targeted molecular markers [26,29]. While these studies have provided important insights into local adaptation, they were generally limited to a small number of loci and did not assess how such trait-associated variation is distributed across natural populations or structured by geographic barriers. Consequently, the extent to which adaptive potential is shaped by coastal configuration and evolutionary connectivity remains poorly understood in many mangrove taxa, including species of the genus Sonneratia.
A genome-wide approach offers a more powerful means of assessing coastal connectivity and identifying distinct evolutionary lineages. This approach enables genetic diversity and population structure to be examined within an explicit spatial and evolutionary framework that cannot be achieved using marker-limited methods. Among molecular markers, single-nucleotide polymorphisms (SNPs) have emerged as the most prevalent marker type in the postgenomic era due to their abundance, stability, and wide genomic distribution [30,31,32,33]. Although individual SNPs contain less information than microsatellites, they are routinely applied in large panels containing thousands of loci, providing greater precision for estimating genetic diversity and enabling more robust assessments of local adaptation [34,35].
High-resolution SNP datasets generated through restriction site-associated DNA (RAD) sequencing allow quantification of genome-wide variation, reveal fine-scale genetic structure, and support evidence-based restoration strategies [36,37]. SNP markers have already been successfully used to evaluate the population genetics of several mangrove species [30,38,39,40,41]. For S. alba and S. caseolaris, however, population structure and genetic diversity have never been examined using genome-wide SNP markers. Such information is essential because restoration programs in Thailand frequently overlook genetic provenance, risking the loss of locally adapted genotypes and potentially reducing long-term ecosystem resilience. Incorporating genomic evidence into mangrove restoration is therefore critical for maintaining adaptive potential under rapid coastal change.
In this study, we evaluated the genetic diversity and population structure of two mangrove species, S. alba and S. caseolaris, along the coastlines of Thailand using high-quality SNP markers generated through RAD sequencing. By applying a genome-wide SNP approach, this study provides the first comparative population genomic assessment of these two species in this region. Our findings elucidate patterns of evolutionary connectivity and spatial genetic structure, with implications for mangrove conservation and restoration efforts.
2. Materials and Methods
2.1. Sample Collection
A total of 107 S. alba and 131 S. caseolaris individuals were sampled from mangrove forests along the Andaman Sea and the Gulf of Thailand coasts (Figure 1; Tables S1 and S2). For S. alba, leaf tissue was collected from 15 provinces in Thailand (Figure 1; Table S1): Chon Buri (CBI; n = 1), Chumphon (CPN; n = 5), Chanthaburi (CTI; n = 10), Krabi (KBI; n = 7), Nakhon Si Thammarat (NST; n = 12), Phuket (PKT; n = 5), Phang-nga (PNG; n = 10), Pattani (PTN; n = 2), Ranong (RNG; n = 6), Rayong (RYG; n = 3), Songkhla (SKA; n = 4), Surat Thani (SNI; n = 14), Satun (STN; n = 12), Trang (TRG; n = 3), and Trat (TRT; n = 13). For S. caseolaris, leaf tissue was collected from 21 provinces (Figure 1; Table S2): Bangkok (BKK; n = 1), Chon Buri (CBI; n = 1), Chachoengsao (CCO; n = 6), Chumphon (CPN; n = 5), Chanthaburi (CTI; n = 10), Nakhon Si Thammarat (NST; n = 14), Narathiwat (NWT; n = 2), Phetchaburi (PBI; n = 5), Phatthalung (PLG; n = 2), Phang-nga (PNG; n = 5), Pattani (PTN; n = 2), Ranong (RNG; n = 10), Rayong (RYG; n = 3), Songkhla (SKA; n = 3), Samut Songkhram (SKM; n = 5), Samut Sakhon (SKN; n = 5), Samut Prakan (SPK; n = 3), Surat Thani (SNI; n = 15), Satun (STN; n = 21), Trang (TRG; n = 5), and Trat (TRT; n = 8). Sites on the Gulf of Thailand include BKK, CBI, CCO, CPN, CTI, NST, NWT, PBI, PLG, RYG, SKA, SKM, SKN, SPK, SNI, PTN, and TRT, whereas Andaman Sea sites include KBI, PKT, PNG, RNG, STN, and TRG. Only young, healthy leaf samples were collected from mature trees, with a minimum spacing of at least 20 m between individuals to minimize resampling of ramets or close relatives. Sample sizes varied among sites according to local population sizes. All samples were collected during 2024–2025. Sampling locations were mapped in QGIS v3.24.2 (Figure 1).
2.2. DNA Extraction, RAD-seq Library Preparation and Sequencing
Genomic DNA was extracted from young leaves using the CTAB method [42]. DNA was further purified with the DNeasy Plant Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions and quantified on a Qubit fluorometer (Thermo Fisher Scientific, Waltham, MA, USA) using the dsDNA BR Assay kit (Thermo Fisher Scientific, Waltham, MA, USA).
RAD-seq libraries were prepared from 1 µg of input DNA per sample using the MGIEasy RAD Library Prep Kit (MGI Tech, Shenzhen, China) following the manufacturer’s instructions. Briefly, genomic DNA was digested with TaqI and fragments were ligated to unique barcoded adapters. The samples were pooled in equimolar amounts and subjected to PCR amplification. Libraries were quantified and quality-checked on a Fragment Analyzer system (Agilent Technologies, Santa Clara, CA, USA) using an NGS Fragment Analysis kit. Sequencing was performed on an MGISEQ-2000RS platform (paired-end 150 bp; MGI Tech, Shenzhen, China) following the manufacturer’s protocol.
2.3. Single-Nucleotide Polymorphisms Identification
Raw sequence data were processed with the Genome Analysis Toolkit (GATK) v4.1.4.1 pipeline [43]. Paired-end, quality-filtered reads from S. alba were aligned to the S. alba reference genome (DDBJ/EMBL/GenBank accession PRJEB8424), and reads from S. caseolaris were aligned to the S. caseolaris reference (Genome Warehouse (GWH) of NGDC accession GWHBCIR00000000) using BWA v0.7.19 -r1273 (https://github.com/lh3/bwa, accessed on 12 July 2025) with default parameters. Sequence alignment map (SAM) files were converted to binary format (BAM), then sorted and indexed using samtools v1.9 [44]. Mapping statistics were also obtained with samtools. SNPs were called using GATK v4.1.4.1 with HaplotypeCaller [43] and filtered using the following thresholds: (i) minor allele frequency (MAF) > 0.05; (ii) depth of coverage between 10× and 200×; and (iii) no missing genotypes. The filtered SNP sets were used for downstream analyses of genetic diversity and population structure in each species.
2.4. Population Genetic Structure Analysis
Genetic clustering for each species was evaluated using a Bayesian model-based approach implemented in STRUCTURE v2.3.4 [45]. We tested K = 1–10 with 20 replicate runs per K under the admixture model with the correlated allele frequencies; each run used a burn-in of 100,000 followed by 1,000,000 MCMC iterations. The optimal K was determined from ΔK [46] and the mean log posterior probability [lnP(D)], as implemented in STRUCTURE Harvester v0.6.94 [47]. Based on the optimal K value, clustering from 10,000 replicates in STRUCTURE was summarized with CLUMPP v1.1.2 [48] to obtain average cluster membership proportions. In addition, principal component analysis (PCA) was used to visualize genetic structure within each species. PCA was performed using the PCA function in R. The first two principal components were selected and plotted using R software v3.3.4 with the library ggplot2 v3.5.0 [49]. To further explore population clustering within each species, unrooted consensus neighbour-joining (NJ) trees were inferred using MEGA11 [50]. A bootstrap consensus tree with 1000 replications was performed. The NJ trees were visualized with iTOL v6 [51].
Analysis of molecular variance (AMOVA) among populations within each species was performed in Arlequin v.3.5 [52] with 10,000 permutations to estimate genetic variance within and among populations based on the results of the clustering analyses. Population differentiation (pairwise FST) among STRUCTURE-inferred clusters and among sampling sites was also estimated with Arlequin v3.5 [52] and visualized in R v3.3.4 using the ggplot2 package v3.5.0 [49]. To reduce bias from small sample sizes, locations represented by a single individual were excluded from the FST analysis.
2.5. Isolation-by-Distance Analysis (Mantel Test)
To determine whether geographic distance explained patterns of genetic differentiation (isolation by distance, IBD) along each coastline in S. alba and S. caseolaris, we conducted a Mantel test in GenAlEx v6.5 [53] with 9999 permutations. Mantel tests were performed separately for the Andaman Sea and Gulf of Thailand populations. Pairwise geographic distances among sampling sites were compared with pairwise genetic distances [FST/(1 − FST)] calculated at the site (population) level.
2.6. Genetic Diversity Analysis
To estimate genetic diversity at the population and species levels, we computed the number of effective alleles (N_e_), Shannon’s information index (I), observed heterozygosity (H_o_), expected heterozygosity (H_e_) and the percentage of polymorphic loci (PPL) for each group using GenAlEx v6.502 [53]. The inbreeding coefficient (FIS) for each population was also calculated in GenAlEx v6.502. Gene flow (N_m_) among groups was approximated as Nm = [(1/FST) − 1]/4 [54]. Polymorphism information content (PIC) values for SNP markers were calculated using PowerMarker v3.25 [55].
3. Results
3.1. Sequencing and SNP Discovery
A total of 107 individuals of S. alba and 131 of S. caseolaris were genotyped using RAD-seq. Sequencing produced 2,001,629,636 and 2,974,679,250 reads for S. alba and S. caseolaris, respectively, of which 89.78% and 92.83% mapped to their respective reference genomes. Average read counts per individual were 18,706,819 for S. alba (Table S1) and 22,707,475 for S. caseolaris (Table S2). Initial SNP calling identified 5,059,036 and 4,696,679 putative loci in S. alba and S. caseolaris, respectively. After applying filtering thresholds, a total of 2664 SNPs for S. alba (Table S3) and 5208 SNPs for S. caseolaris (Table S4) were retained for downstream analyses. Polymorphic information content (PIC) was estimated for each SNP to quantify marker informativeness across 107 S. alba and 131 S. caseolaris individuals. In S. alba, PIC values ranged from 0.09 to 0.58 (mean = 0.19), whereas in S. caseolaris, PIC values ranged from 0.27 to 0.38 (mean = 0.34) (Figure S1).
To further characterize genome-wide patterns of genetic variation, we examined the chromosomal distribution and functional positions of RAD-seq SNPs in both species. In S. alba, SNPs were identified across all chromosomes, with the number of variants per chromosome ranging from 144 to 335 (Table S5). Similarly, in S. caseolaris, SNPs were detected on all chromosomes, with variant counts ranging from 281 to 636 per chromosome (Table S6). To explore patterns of SNP distribution in both species, SNP frequency was plotted at 1 Mb intervals along each chromosome. SNP density across the genome ranged from 0 to 50 SNPs per megabase (Mb), with an average density of approximately one SNP per 75 kb in S. alba and one SNP per 36 kb in S. caseolaris. Although the number of SNPs varied among chromosomes and between species, both datasets exhibited broad genome-wide coverage rather than concentration on a limited number of chromosomes (Figure S2). Regions of chromosomes lacking SNPs generally corresponded to areas where no RAD-seq reads were recovered, rather than indicating an absence of polymorphism.
Functional annotation of SNPs revealed similar genomic distributions in both species. In S. alba, most SNPs were located in downstream (34.41%) and upstream (30.49%) regions, followed by exonic (13.99%) and intronic (10.76%) regions, with smaller proportions occurring in intergenic regions and splice-related sites. A comparable pattern was observed in S. caseolaris, where downstream (32.41%) and upstream (32.15%) regions contained the largest fraction of SNPs, followed by intronic (10.35%) and exonic (11.43%) regions. Because SNP calling was performed independently for each species using species-specific reference genomes, direct positional comparisons of SNPs between S. alba and S. caseolaris were not conducted. Accordingly, comparisons focused on overall genome-wide patterns of SNP distribution and functional category composition rather than one-to-one positional correspondence between species.
The RAD-seq approach applied in this study was not designed to specifically target previously reported candidate genes associated with adaptive traits. Consequently, direct detection of such trait-associated variants could not be confirmed, and coverage of these genomic regions cannot be inferred from the present dataset.
3.2. Population Genetic Structure
To understand the population structure of S. alba and S. caseolaris in Thailand, three different approaches—STRUCTURE, PCA and a neighbour-joining (NJ) tree—were applied. All analyses used the SNP genotype sets of 2664 loci for S. alba and 5208 for S. caseolaris. In STRUCTURE, the number of clusters was determined using the ΔK method [46]. The ΔK distribution showed clear modes at K = 3 for S. alba and at K = 2 for S. caseolaris (Figure S3). Under the Evanno criterion, the STRUCTURE results provided the strongest support for three genetic clusters in S. alba and two clusters in S. caseolaris (Figure 2). In S. alba, at K = 3, ancestry coefficients revealed a clear coastal partition, with one cluster predominating along the Andaman Sea coast and another along the Gulf of Thailand coast. A third cluster appeared primarily as admixture across several sites. Populations from KBI, PKT, PNG, RNG, and STN (Andaman Sea) together with TRT and NST (Gulf of Thailand) formed one genetic cluster and shared inferred ancestry with individuals from both coasts (Figure 2A). For S. caseolaris, most individuals from the same geographic region were assigned to the same genetic cluster (Figure 2B). Cluster 1 (orange) was dominated by Andaman Sea samples (PNG, RNG, STN, TRG), whereas Cluster 2 (green) comprised mainly Gulf of Thailand samples (BKK, CCO, CPN, CTI, NST, NWT, PBI, PLG, PTN, RYG, SKA, SKM, SKN, SNI and TRT). Some individuals showed mixed ancestry, and a few, particularly those from PNG, grouped with the opposite coast, indicative of admixture or recent gene flow. Consistent with the STRUCTURE results, both the PCA and the NJ tree revealed a pronounced genetic divide between the Andaman and Gulf of Thailand coasts (Figure 3), with each cluster largely confined to one coast. The first two principal components explained 55.65% and 32.34% of the total genetic variance in S. alba and S. caseolaris, respectively. The NJ trees of both species also revealed two major clades corresponding to the two coastal regions, consistent with the STRUCTURE and PCA results (Figure 3).
3.3. Genetic Differentiation of Populations
We quantified genetic differentiation (FST) among STRUCTURE-defined subpopulations to assess relationships among groups. Following standard thresholds [56], FST values of 0.00–0.05 indicate low differentiation, 0.05–0.15 indicate moderate differentiation, and > 0.15 indicate high differentiation. Genetic differentiation among STRUCTURE-defined populations was high in both species (S. alba: FST = 0.364, p < 0.001; S. caseolaris: FST = 0.321, p < 0.001). The mean number of migrants per generation (N_m_), estimated from FST as N_m_ = [(1/FST) − 1]/4, was 0.437 for S. alba and 0.528 for S. caseolaris. Values of N_m_ < 1 indicate limited gene flow, consistent with the high FST values, and together these results point to substantial population divergence in both species. Pairwise FST estimates indicated high differentiation, ranging from 0.296 to 0.412 for S. alba and 0.321 for S. caseolaris (two-group comparison; Table S7). In S. alba, the greatest divergence occurred between subpopulation 1 and subpopulation 2 (FST = 0.412). AMOVA revealed significant genetic differentiation among STRUCTURE-defined populations in both species (p < 0.01). In S. alba (K = 3), 36.43% of the variance was partitioned among populations and 63.57% within populations. In S. caseolaris (K = 2), 32.15% of the variance occurred among populations and 67.85% within populations (Table 1). Overall, population structure is pronounced in both species, with S. alba exhibiting stronger differentiation than S. caseolaris.
Population differentiation (pairwise FST) among sampling locations was also estimated. For S. alba, pairwise FST ranged from 0.002 to 0.720 (Figure 4A and Table S8). The largest values were generally observed for between-coast comparisons of sampling sites, such as RNG and SKA, whereas the smallest values occurred within coasts, including RYG–TRT and TRG–KBI. For S. caseolaris, pairwise FST ranged from 0.007 to 0.651 (Figure 4B and Table S8). High differentiation was concentrated in between-coast comparisons of sampling sites, such as RNG–PTN, whereas within-coast pairs, such as PBI–SKM, showed the lowest values. Mean pairwise FST by coast pairing showed that, for S. alba, values were 0.075 within the Andaman Sea, 0.173 within the Gulf of Thailand, and 0.435 between coasts. For S. caseolaris, values were 0.348 within the Andaman Sea, 0.209 within the Gulf of Thailand, and 0.465 between coasts. In both species, the between-coast mean exceeded the within-coast means, consistent with strong coastal structure.
3.4. Correlation Analysis Between Genetic Distance and Geographic Distance
In order to explore the spatial genetic structure of populations, the Mantel test is commonly used to assess whether the species conform to the model of isolation by distance (IBD) by evaluating the correlation between genetic and geographic distances. Patterns of IBD differed between S. alba and S. caseolaris (Figure 5). For S. alba, Mantel tests showed no significant correlation between pairwise genetic distances and coastal geographic distances along either coastline. The Andaman Sea populations exhibited a weak, non-significant positive trend (R^2^ = 0.152; p = 0.066), while the Gulf of Thailand populations showed no relationship (R^2^ = 0.003; p = 0.353). These results indicate that geographic distance is not a major driver of genetic differentiation in S. alba across the sampled sites. In contrast, S. caseolaris exhibited clear evidence of IBD along both coasts. A strong and significant correlation was detected in the Andaman Sea populations (R^2^ = 0.728; p = 0.044), and a moderate but significant correlation was also observed among Gulf of Thailand populations (R^2^ = 0.247; p = 0.001). These results suggest that genetic divergence in S. caseolaris increases with coastal distance, implying more spatially restricted propagule dispersal and stronger geographic structuring relative to S. alba.
3.5. Genetic Diversity
Diversity statistics were computed for two coastal groupings (Andaman Sea vs. Gulf of Thailand) in each species (Table 2). For S. alba, mean values across the two coasts for the effective number of alleles (N_e_) and Shannon’s index (I) were 1.335 and 0.367, respectively. The Andaman Sea group showed slightly higher diversity than the Gulf of Thailand group (Andaman Sea: I = 0.382, H_e_ = 0.232, H_o_ = 0.196; Gulf of Thailand: I = 0.353, H_e_ = 0.213, H_o_ = 0.150). Overall, H_o_ (0.173) was lower than H_e_ (0.223), consistent with heterozygote deficiency. PPL was high and comparable between coasts (Andaman Sea 99.66%, Gulf of Thailand 99.89%). For S. caseolaris, mean N_e_ and I across coasts were 1.726 and 0.589, respectively. The Andaman Sea group exhibited slightly lower diversity than the Gulf of Thailand group (Andaman Sea: I = 0.554, H_e_ = 0.377, H_o_ = 0.303; Gulf of Thailand: I = 0.624, H_e_ = 0.434, H_o_ = 0.277). Overall, H_o_ (0.290) was lower than H_e_ (0.406), indicating heterozygote deficiency. PPL was high and comparable between coasts (Andaman Sea 98.21%, Gulf of Thailand 100.00%). The inbreeding coefficient (F_IS_) was positive on both coasts in both species. Genetic diversity was lower in S. alba than in S. caseolaris (mean H_e_ = 0.223 vs. 0.406; N_e_ = 1.335 vs. 1.726), with only minor differences between coasts within each species.
4. Discussion
Understanding genetic diversity and population structure is essential for developing informed conservation strategies for mangrove genetic resources. In this study, we used SNP markers to evaluate the genetic diversity and population structure of S. alba and S. caseolaris from the coastal range of Thailand. The observed population stratification offers insight into how geographic configuration and species-specific ecological traits influence evolutionary processes in mangrove systems. STRUCTURE analysis separated S. alba populations into three groups (one showing admixture), whereas S. caseolaris populations were divided into two major genetic clusters. Importantly, the inferred population structure corresponded closely to the geographical origins, with clear separation between the Andaman Sea and the Gulf of Thailand coasts. Both PCA and the NJ tree supported this pattern, indicating that coastal configuration is a dominant driver of genetic differentiation in both species. Interestingly, one genetic cluster of S. alba showed genetic admixture between the two coastal regions, while such an admixture group was not observed in S. caseolaris. This contrast may reflect differences in ecological niche and dispersal dynamics between the two species. S. alba typically occupies exposed seaward fringe habitats and produces buoyant propagules that may occasionally disperse over longer distances. In addition, anthropogenic translocation associated with mangrove restoration programs, where seedlings are moved across regions to maximize planting success, may have contributed to the observed admixture in S. alba populations [57]. In contrast, S. caseolaris predominantly occurs in sheltered estuarine environments, where propagule dispersal is likely more localized, resulting in stronger genetic isolation between coastal regions.
High levels of genetic differentiation were observed between subpopulations of S. alba (FST = 0.364, p < 0.001) and S. caseolaris (FST = 0.321, p < 0.001), whereas pairwise FST values within the same coastal region were relatively low. This pattern indicates ongoing gene exchange among geographically proximate populations but limited connectivity across the two coasts. The Malay Peninsula likely acts as a major land barrier that restricts oceanic circulation and prevents the dispersal of sea-drifted propagules between the Andaman Sea and the Gulf of Thailand. Similar east–west genetic breaks have been reported in several mangrove species, including Rhizophora mucronata [58], Rhizophora apiculata [40,58], Bruguiera parviflora [38], Bruguiera cylindrica [59], Bruguiera gymnorhiza [37], and Ceriops tagal [39], supporting the Malay Peninsula barrier hypothesis [60]. The relatively low estimates of gene flow (N_m_ = 0.437 in S. alba and N_m_ = 0.528 in S. caseolaris; Table 1) are consistent with this interpretation.
Isolation-by-distance (IBD) analyses further revealed species-specific patterns of spatial genetic structure. No significant correlation between genetic and geographic distance was detected in S. alba, suggesting that spatial separation alone does not explain its genetic structure. In contrast, S. caseolaris exhibited significant IBD patterns along both coasts, indicating that gene flow in this species is more sensitive to geographic distance. This pattern is consistent with IBD signals reported in other mangrove species, such as Rhizophora mangle [61]. The genetic structure may be influenced by multiple factors, including the Malay Peninsula barrier, geographic distance, ocean currents, sea level changes, climatic variation, and other environmental processes that interact to shape the spatial genetic patterns of mangrove populations in Thailand [27,60,61,62,63,64,65].
While these results provide robust insights into population structure and evolutionary connectivity, it is important to clarify the scope, strengths, and limitations of the genomic approach applied in this study. The RAD-seq approach employed here was designed to efficiently capture genome-wide SNP markers suitable for population-level inference in non-model species, enabling characterization of chromosomal SNP distributions and broad patterns of genomic variation across populations. This approach allowed assessment of SNP recovery across chromosomes and classification of variants according to their genomic positions, thereby providing additional biological context for the observed population genetic structure. However, RAD-seq also has inherent limitations. Because SNP discovery is restricted to regions flanking restriction enzyme cut sites and relies on species-specific reference genomes, the approach is not optimized for comprehensive functional annotation, fine-scale linkage disequilibrium analyses, or direct cross-species comparisons of homologous variants. In addition, this restriction-site–dependent sampling limits the ability to comprehensively capture known adaptive loci previously identified through candidate gene approaches. Consequently, while the present study provides genome-wide insights into population structure and evolutionary connectivity, direct assessment of trait-associated genetic variation will require complementary approaches integrating population genomics with targeted functional analyses. Nevertheless, the RAD-seq dataset generated in this study provides a robust framework for detecting evolutionary subdivision and assessing connectivity among mangrove populations, and represents a valuable resource for future studies that aim to link population genomic patterns with functional and adaptive variation.
The levels and patterns of genetic variation in S. alba and S. caseolaris revealed low genetic diversity across species and coastal regions. S. alba showed lower genetic diversity than S. caseolaris (mean H_e_ = 0.223 vs. 0.406; N_e_ = 1.335 vs. 1.726), consistent with previous studies reporting reduced genetic variability in S. alba relative to other mangrove taxa [24]. This may reflect its more restricted ecological niche in exposed seaward zones, which makes the species more vulnerable to demographic bottlenecks and environmental disturbances [66]. In contrast, S. caseolaris typically occupies sheltered estuarine habitats, which can support larger and more stable population sizes [67]. Our findings are consistent with reports of low genetic diversity in S. alba populations across Sabah, Malaysia (H_e_ = 0.21; Hₒ = 0.21; [68]) and across the Indo–West Pacific (H_e_ = 0.28; Hₒ = 0.26; [27]), although some studies have reported slightly higher values [63]. For S. caseolaris, the diversity observed in Thailand was also low and comparable to that reported for populations in Vietnam (I = 0.447; h = 0.300) [28]. Similar patterns of low genetic diversity have also been documented in other mangrove species [69,70,71,72,73,74,75], suggesting that reduced genetic variation may be a common feature of mangrove ecosystems subject to strong environmental filtering and anthropogenic pressure.
Low genetic diversity in S. alba and S. caseolaris suggests increased vulnerability to multiple biological and environmental stressors. Breeding systems, vegetative reproduction, and propagule dispersal patterns can also influence genetic diversity within mangrove populations [69,76,77]. Additionally, populations of both species are threatened by complex coastal geomorphology and a range of human-induced pressures, including urbanization, wood harvesting, conversion to agriculture and aquaculture, and tourism development [18,78,79]. These factors may accelerate habitat fragmentation and lead to further loss of genetic diversity in S. alba and S. caseolaris in Thailand. The particularly low diversity observed in S. alba demonstrates the need for conservation actions focused on maintaining gene flow, protecting remnant populations, and preventing further fragmentation. From a broader biological and conservation perspective, the pronounced genetic subdivision observed between the Andaman Sea and Gulf of Thailand populations indicates limited evolutionary connectivity between coastal regions. Conservation planning should therefore account for regional genetic differentiation when implementing mangrove restoration programs. Maintaining populations within their respective coastal zones during propagule sourcing may help preserve distinct genetic lineages and reduce the risk of disrupting locally adapted populations. In situ conservation appears appropriate for both species, while carefully guided augmentation using genetically informed sources from high-diversity populations may enhance genetic variation in depleted populations. The use of SNP-based genomic information to guide such management decisions can help prevent unintentional genetic homogenization across regions and support the long-term adaptive potential of mangrove ecosystems under ongoing environmental change.
5. Conclusions
This is the first study to investigate the genetic diversity and population structure of S. alba and S. caseolaris across the coastal range of Thailand using genome-wide SNP markers. Population structure analyses, PCA, and NJ trees showed that S. alba populations were clustered into three major groups, whereas S. caseolaris populations were clustered into two major groups. These clusters exhibited a clear geographical pattern corresponding to the Andaman Sea and Gulf of Thailand coasts, suggesting that genetic dispersal in both species is strongly influenced by the geographic barrier of the Malay Peninsula. AMOVA and pairwise FST analyses indicated relatively high genetic differentiation between populations, with genetic variation partitioned mainly within populations. Overall, low levels of genetic diversity were observed in both species. Together, these findings provide genome-wide insights into the evolutionary divergence and coastal connectivity of S. alba and S. caseolaris in Thailand. The clear regional subdivision observed in both species indicates that populations from the two coasts represent distinct evolutionary lineages. Maintaining these lineages in restoration and reforestation efforts will help preserve their adaptive potential, particularly under accelerating coastal change. Therefore, matching planting materials to their native coastal regions may help strengthen the long-term resilience of Thailand’s mangrove forests.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Sremongkontip S. Hussin Y.A. Groenindijk L. Detection C. Detecting changes in the mangrove forests of southern Thailand using remotely sensed data and GIS Int. Arch. Photogramm. Remote Sens.200033567574
- 2Kathiresan K. Dagar J.C. Mangroves and Associated Flora: Prospects for Utilization in Coastal Agriculture Halophytes Vis-à-Vis Saline Agriculture: Perspectives and Opportunities for Food Security Dagar J.C. Gupta S.R. Kumar A. Springer Nature Singapore Singapore 20246795
- 3Jennerjahn T.C. Ittekkot V. Relevance of mangroves for the production and deposition of organic matter along tropical continental margins Naturwissenschaften 200289233010.1007/s 00114-001-0283-x 12008969 · doi ↗ · pubmed ↗
- 4Macreadie P. Anton A. Raven J. Beaumont N. Connolly R. Friess D. Kelleway J. Kennedy H. Kuwae T. Lavery P. The future of Blue Carbon science Nat. Commun.201910399810.1038/s 41467-019-11693-w 31488846 PMC 6728345 · doi ↗ · pubmed ↗
- 5Zhang K. Liu H. Li Y. Xu H. Shen J. Rhome J. Smith T.J.III The role of mangroves in attenuating storm surges Estuar. Coast. Shelf Sci.2012102112310.1016/j.ecss.2012.02.021 · doi ↗
- 6Chanda S. Mangrove Forest Ecosystem: Services, Conservation, Restoration and Carbon Finance Forests and Climate Change: Biological Perspectives on Impact, Adaptation, and Mitigation Strategies Singh H. Springer Nature Singapore Singapore 2024625651
- 7Leung J.Y.S. Tam N.F.Y. Influence of plantation of an exotic mangrove species, Sonneratia caseolaris (L.) Engl., on macrobenthic infaunal community in Futian Mangrove National Nature Reserve, China J. Exp. Mar. Biol. Ecol.20134481910.1016/j.jembe.2013.06.006 · doi ↗
- 8Kathiresan K. Bingham B.L. Biology of mangroves and mangrove Ecosystems Advances in Marine Biology Academic Press Cambridge, MA, USA 2001 Volume 4081251