Gut Microbiome Variations in Herring Gulls (Larus argentatus) from Different Environments in the United Kingdom
Wai Tung Kan, Samantha A. Siomko, Nicola J. Rooney, Paul Wigley

TL;DR
This study explores how the gut microbiome of herring gulls varies across different UK environments, finding differences linked to urbanization and captivity.
Contribution
The study provides new insights into how environmental factors influence gut microbiome diversity in wild and captive herring gull populations.
Findings
Captive gulls and those in Bristol had higher gut microbiome diversity compared to urban and suburban gulls.
Urban gulls showed higher abundance of Ligilactobacillus and lower abundance of Streptococcus compared to suburban gulls.
The Liverpool population had a high proportion of Mycoplasma, potentially indicating avian mycoplasmosis.
Abstract
In recent years, scientists have been actively seeking non-invasive approaches to monitoring the health and welfare of wildlife. The Herring Gull is a highly adaptable species, which offers an excellent opportunity to study various potential environmental influences, including captivity, level of urbanisation, habitat types and geographical location. Here, we utilised faecal samples from different herring gull populations to analyse variation in their gut microbiome—the bacterial community residing in the guts. Populations sampled for this study include Bristol, Gloucester, Weston Super-Mare, Portishead, Hinkley Point, Steepholm Island, Hereford Wildlife Rescue Centre, Liverpool, Swansea and West Kirby. Significant differences were found between individual populations. Higher gut microbiome diversity (number of bacterial species) was identified in the captive population and in gulls…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
 Figure 1
Figure 1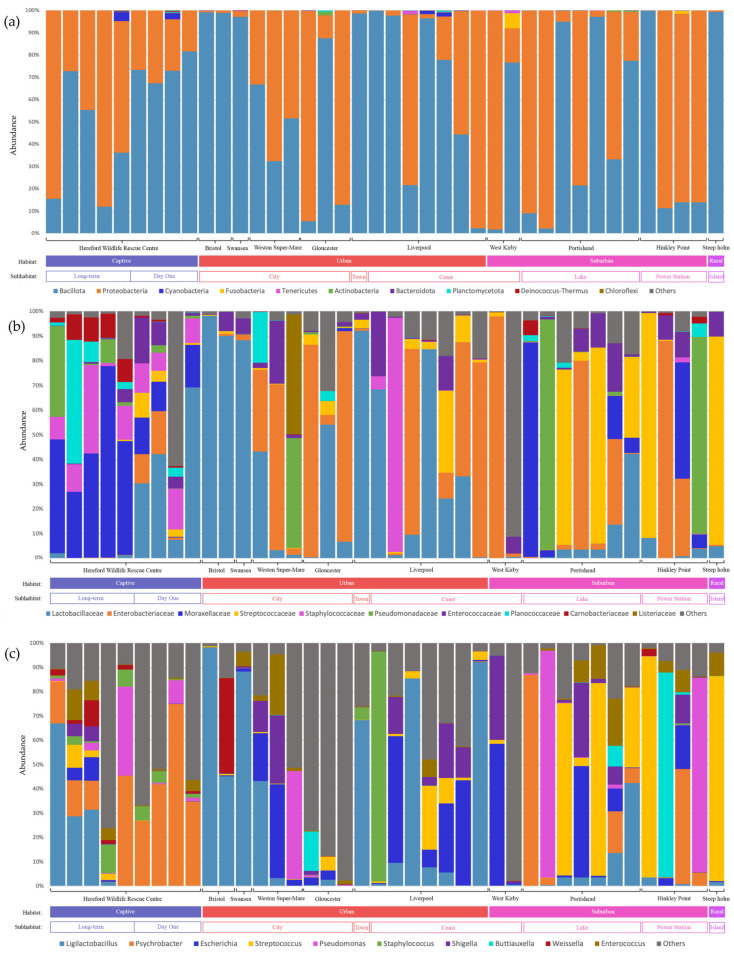 Figure 2
Figure 2- —SWBio BBSRC Doctoral Training Partnership
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsGut microbiota and health · Animal Nutrition and Physiology · Microbial infections and disease research
1. Introduction
With the ongoing growth of human populations and the expansion of cities, natural environments are inevitably altered for agricultural, industrial and other purposes. Related consequences include the destruction and degradation of natural habitats and subsequent environmental pollution, climate change and rapid species extinction [1]. Also, numerous studies on the ecosystem and wildlife population have revealed negative effects of urbanisation [2]. It is, therefore, important to consider the possible impact of human activities on the internal health of wild animals, which is often overlooked and understudied. This is especially true for species adapted to live alongside humans and highly mobile species, like birds, as their foraging range often overlaps with human settlements. However, even species that tend to remain in their natural habitats suffer unavoidable changes like habitat fragmentation, landscape transformations and pollution.
The microbiome can be influenced by habitat disturbances [3,4], and analysis of the gut microbiome as a way to understand animal health has been gaining attention from researchers [5]. These bacterial communities inhabiting the host gastrointestinal (GI) environment, namely the gut microbiome, are found to aid in physiological functions like digestion, nutrient absorption and immunity, as well as cognitive abilities and behavioural processes [6]. Although current research efforts focus mostly on mammals and domesticated birds, it is found that both genetic factors [7,8] and external factors, like diet [9,10] and environment [11,12], contribute to wild avian gut microbiome diversity and composition. With over 10,000 wild avian species inhabiting diverse ecological niches, their gut microbiome health is vital for individual health and welfare, which is, in turn, essential for them to sustain their ecological roles. Moreover, given the migratory nature of most avian species, their wide geographical distribution and their potential as disease reservoirs [13], further understanding of their gut health could aid in disease control efforts.
In the few studies of how differences in environment contribute to wild birds’ gut microbiome changes, several environmental factors have been shown to have significant influences. The effects of factors including habitat [14,15], climate [16], diet [9,10], pollution [17,18] and captivity [19] have been assessed and revealed, mostly by comparative studies of different populations, while some incorporated experimental approaches. In one study of Swainson’s thrushes (Catharus ustulatus) and grey catbirds (Dumetella carolinensis), researchers compared the spring migrants with the fall migrants and found greater microbial compositional differences with seasonality than between species [20]. Hence, environmental factors appeared to have outweighed that of their host genetics for these two species. Other comparative studies on single species in different environmental conditions have also seen differences across various parameters, like alpha diversity, beta diversity and composition. For instance, the percentage of urban land coverage was shown to be associated with shifts in gut microbiome composition in American white ibises (Eudocimus albus) [13].
Most studies on avian species focus on passerines, while other species, like water birds, are largely unstudied [21]. The Herring Gull (Larus argentatus), a widely spread species across different habitats with abundant populations in the UK and Europe, is an ideal candidate for exploring the potential effect of environmental differences on the gut microbiome. This species is highly adapted to urbanised areas, living alongside humans, and along with some other sea birds, is on the UK Red List due to its rapid decline in number [22]. Comparative analysis between populations in locations with different urbanisation levels could provide insight into anthropogenic impact on their gut health [22]. Furthermore, urban gulls are considered a risk in the transmission of antimicrobial-resistant and zoonotic bacteria carried within the intestinal tract [23]. This study aims to compare and identify differences in the gut microbiome diversity and composition across Herring Gull populations living in locations with different degrees of urbanisation, in different geographic areas and also in captive settings.
2. Materials and Methods
2.1. Sample Collection
We sampled nine wild and one captive (Hereford Wildlife Rescue Centre HWRC) locations across Western UK (Figure 1). We obtained between 2 and 12 samples per site. At each site, between 1 and 10 h were spent observing gulls, resulting in a total of about 44 sampling hours. A total of 69 samples were collected between 25 June 2024 and 26 July 2024 and categorised by “Sites”, “Habitat” and “Sub-Habitat” according to the location of collection (Figure 1; Table 1). All wild samples were collected by direct observation of defecating Herring Gull individuals and immediate swabbing of faecal material using sterile collection swabs. Herring Gulls in the HWRC were separated into individual crates and were left alone for about 10–20 min prior to routine health checks. Faecal samples were collected from the crates using a sterile collection swab after returning the individuals to their enclosure. Juvenile individuals are indistinguishable between Herring Gulls and Lesser Black-backed Gulls (Larus fuscus), so sampled individuals seen alongside adult Herring Gulls were assumed to be juveniles of that species. Care was taken to avoid the white part of the droppings, which contains uric acid which can inhibit DNA extraction [24]. Faecal samples were placed in a sterile tube and stored in an insulated cool bag with ice blocks immediately after collection. All samples were placed in commercial freezers within 24 h of collection, then moved to −20 °C and finally −80 °C for long-term storage within a month until extraction.
2.2. DNA Extraction
DNA was extracted using the ZymoBIOMICS DNA Miniprep Kit (Zymo Research, Irvine, CA, USA) from 200 mg of thawed faecal material according to manufacturer’s protocol. The resulting DNA samples were eluted in 100 μL of ZymoBIOMICS^TM^ DNase/RNase Free Water. Extracted DNA was stored at 4 °C until sequencing. To quantify the DNA concentration, the Qubit dsDNA HS fluorometric kit (Thermo-Fisher Scientific, San Jose, CA, USA) was used. Considering the quality of the DNA samples, 46 out of 69 samples were chosen for PCR and sequencing.
2.3. Library Preparation for Nanopore Sequencing
The 16S Barcoding Kit 24 V14 (Oxford Nanopore Technologies, Oxford, UK) was used for 16S amplicon sequencing according to the manufacturer’s protocol. Samples were standardised to 10 ng DNA, and the entire 16S rRNA gene was amplified by PCR using primers 27F (5′-AGAGTTTGATCCTGGCTCAG-3′) and 1492R (5′-TACGGYTACCTTGTTACGACTT-3′) and LongAmp Taq 2x Master Mix (New England Biolabs, Ipswich, MA, USA).
The Agilent SureCycler 8800 was used for PCR and programmed as described in the barcoding kit. DNA concentration of the resulting amplified samples was quantified using the Qubit dsDNA HS fluorometric kit (Thermo-Fisher Scientific, San Jose, CA, USA), and all barcoded samples were pooled in varying volumes to contain a maximum of 75 ng DNA, according to individual concentration.
2.4. Nanopore Sequencing
The MinION flow cells by Oxford Nanopore Technology, Oxford, UK were primed and loaded for sequencing following the manufacturer’s protocol for the corresponding kit (16S Barcoding Kit 24 V14). Sequencing was performed for 74 h with the MinKNOW application (24.02.16) using the default settings with the function “Barcode Balancing” to account for uneven DNA concentrations across samples. A positive control mock microbial community was not included in this study; therefore, taxonomic assignments were interpreted in the context of known limitations of nanopore-based 16S sequencing and species-level analysis was avoided to reduce uncertainty. Negative sequencing control did not contain a significant number of reads. Basecalling was carried out on the GridION using Oxford Nanopore’s basecalling software (Dorado 7.3.11). Basecalled FASTQ files were subsequently analysed using the Epi2Me platform.
2.5. Taxonomy Assignment
EPI2ME software version 5.1.14 (Oxford Nanopore Technologies, Oxford, UK) was used to analyse the FASTA files with the wf-16s workflow and Minimap2 classifier. Reports were generated for alpha diversity metrics and OTU taxonomy abundance classified at the phylum to genus levels. Samples with the number of reads drastically lower (<1000) than the rest of the samples are not included in the data analysis, reducing the sample size to n = 40. Taxonomic richness and diversity metrics were calculated from unrarefied abundance tables generated by EPI2ME, and richness values were interpreted in a relative framework given sufficient sequencing depth across samples (Figure S1).
2.6. Data Analysis and Statistics
The alpha diversity metrics, including Pielou’s evenness, Berger-Parker index, Simpson’s Diversity Index, Inverse Simpson index, Shannon’s diversity index, total OTU count per sample, richness per sample and effective number of species, were considered. To analyse the microbial composition, abundance tables at the phylum, family and genus levels were devised. The taxa chosen for analysis were based on abundance and possible relevance (i.e., reported taxa related to external change from previous research papers).
Samples were categorised into “Sites”, “Habitat” and “Sub-Habitat” (Table 1). “Sites” refers to which sampling site the samples were from. Group definitions under the “Habitat” and “Sub-Habitat” categories are shown in Table 1. The groups “Rural”, “Town”, “Swansea” and “Steep-holm Island” were, however, excluded from any data analysis due to their extremely small group size (n = 1). However, all of the samples from these groups are still presented in bar charts. Apart from the sample of “Steepholm Island” and “Rural”, which is the same sample, other samples are still included in at least one of the ANOVA tests. For example, although “Swansea” is too small to be analysed as a “Site”, it still belonged to “Urban” when ANOVA tests were performed on “Habitats”. An additional category of “Age” is also applied to captive individuals to assess the age effect on the gut microbiome.
All statistical analyses were performed using R version 4.3.2. We tested for differences in alpha diversity metrics and taxa abundances between groups according to categories using ANOVA (Tables S1, S3, S5 and S7). When tested significant (p ≤ 0.05) or close to significant (p ≤ 0.10), post hoc Tukey’s Honest Significant Difference (HSD) test was performed with base R (Tables S2, S4, S6 and S8). The prevalence of Mycoplasma was calculated by considering the percentage of individual samples consisting of Mycoplasma. To visualise the microbial composition by phylum and family, stacked bar charts were created using Excel.
3. Results
Most of the results from ANOVA tests showed nonsignificant differences in alpha diversity metrics (Table 2 and Table S7). When comparing groups of “Sites”, there was a significant difference in the Inverse Simpson index. Further Tukey’s HSD tests suggested that “Bristol” had a significantly higher Inverse Simpson index than all the other sites (p < 0.05), with “West Kirby” being the most similar to “Bristol” (p = 0.0298) and “HWRC” being the most dissimilar (p = 0.0017) (Table S8).
When comparing “Habitats”, significant differences were identified in Shannon’s diversity index and the Effective number of species. Tukey’s HSD test reveals a significantly higher Shannon’s diversity index in “Captive” compared to both “Urban” (p = 0.0398) and “Suburban” (p = 0.0473). When considering the Effective number of species, the “Captive” population exhibited diversity when compared to the “Suburban” population (p = 0.028) only.
Bacillota and Proteobacteria showed the highest relative percentage of the total microbiome community at the phylum level (Figure 2a), but differences were much more pronounced at the family and genus level (Figure 2b,c).
Out of the seven phyla analysed (Table 3, Tables S1 and S2), significant differences were found between “Sites” in Bacillota (F = 2.468; p = 0.0399) and Fusobacteriota (F = 2.835; p = 0.0216). “West Kirby” displayed a significantly higher abundance of Fusobacteriota than did “Liverpool” (p = 0.0166), “Hereford Wildlife Rescue Centre” (p = 0.008), “Gloucester” (p = 0.0306), “Portishead” (p = 0.01) and “Weston Super-mare” (p = 0.0306). For Bacillota, however, there was no significant difference despite an observable trend for a higher abundance in the “Bristol” population (0.09 > p > 0.05).
Staphylococcaceae was the only family that differed significantly between “Habitat” (F = 4.533; p = 0.0175) with a higher abundance in “Suburban” than “Urban” population (p = 0.0226) and between “Sub-Habitat” (F = 2.592; p = 0.0446; Table 3, Tables S3 and S4). Although the “Lake” population showed a trend of higher abundance than that of “City” (p = 0.0501) and “Coast” (p = 0.0582), Tukey’s HSD test showed that no two sub-habitats differed significantly (Table S4). Since “Site” differences were close to significant (p = 0.0657), Tukey’s HSD test was carried out for the abundance differences in the Lactobacillaceae family. The “Gloucester” population had a higher abundance than “Liverpool” (p = 0.0228), “Portishead” (p = 0.0357) or “HWRC” (p = 0.0306) (Table S4).
When investigating the inter-group difference in abundance at the genus level, we found several significant differences across the “Sites”, “Habitats” and “Sub-Habitats” categories (Tables S5 and S6). Differences between “Sites” were identified in the genera Bacillus (F = 2.557; p = 0.0343), Ligilactobacillus (F = 2.736; p = 0.0254), Klebsiella (F = 5.777; p = 0.000266) and Mycoplasma (F = 2.49; p = 0.0178). Tukey’s HSD tests showed a significantly more abundant Bacillus bacteria community in the “Gloucester” site than in “HWRC” (p = 0.0113), “Hinkley Point” (p = 0.033) and “Liverpool” (p = 0.0244) (Table S6). The “Gloucester” population also showed a higher abundance of Klebsiella compared to all other “Sites” (0.0007 < p < 0.006), with a highly significant difference from “HWRC” (p = 0.00007). The “Bristol” site had a higher abundance of Ligilactobacillus than “Gloucester” (p = 0.0497), “Hinkley Point” or “Liverpool”. It is noteworthy that Mycoplasma was present in only 12 out of 40 samples (30%), with individuals from Liverpool making up most of those (7 individuals out of 8).
Among different “Habitats”, the abundance of Ligilactobacillus varied significantly (F = 3.466; p = 0.042), as did Streptococcus (F = 3.475; p = 0.0417) (Table S6). The “Urban” samples had a significantly higher abundance of Ligilactobacillus than the “Suburban” (p = 0.0387), whilst the “Suburban” population exhibited a nonsignificant trend towards higher Streptococcus than the “Urban” (p = 0.05001).
Genera abundances of Acinetobacter (F = 2.684; p = 0.039) and Mycoplasma (F = 2.557; p = 0.0469) also varied significantly between different “Sub-Habitats”(Table S6). The “Long-term” captive population had a more abundant Acinetobacter community than the “City” (p = 0.0424) and “Coast” populations (p = 0.0350), whilst Tukey’s HSD results found no significant difference in Mycoplasma between Sub-Habitats.
4. Discussion
This study aimed to identify the possible differences in gut microbiome diversity and composition between Herring Gull populations from various environments. Although the gut microbiome of all samples was dominated by Bacillota and Proteobacteria, we found differences in alpha diversity indexes and the microbial composition between sites and habitats. These findings align with most other studies around the impacts of external factors on avian gut microbiome, including habitat types, diet [9,10], pollution [25,26], and captivity [27]. Our results suggest the possible impact of captivity, urbanisation and geographical differences on avian gut microbiome.
4.1. The Effect of Captivity
When comparing the sequencing results of captive Herring Gulls with other free-roaming populations from different sites, we found a significantly higher Shannon’s diversity in the captive population than in urban and suburban populations. As one of the most used and effective diversity indexes, this finding suggests that Herring Gulls under captivity have a higher microbiome diversity. Another study also reported a similar result comparing a group of Common Kestrels (Falco tinnunculus) prior to and during captivity [27]. However, in contrast, a study of oriental white storks (Ciconia boyciana) [19] saw a less diverse gut microbiome in captive compared to wild birds [19]. Differences in diet, enclosure cleanliness, and size and other variables could explain the contrasting results. The association between diet and the wild avian gut microbiome is relatively well-established [9,21,28,29,30]. At our captive sampling location, the HWRC, gulls were fed a mix of dog food, chicken and fresh fish. This diet is likely to be higher in protein and dietary fibre than that of the Herring Gulls living near human settlements [31] who scavenge on human leftovers like chips, bread and other processed foods, which are low in MACs (microbiota-accessible carbohydrates), complex carbohydrates found in dietary fibre [32]. MACs are crucial carbon and energy sources for distal gut microbiota, and hence influential for the microbial community [32], which could explain the higher diversity of their gut microbiome in captivity. Similarly severe and long-lasting reductions in gut microbial diversity were observed in many studies on mammals with a low-MACs diet (processed food, high in fat and low in fibre) [32].
A higher abundance of Acinetobacter was also identified in the “Long-term” group, which are rescued gulls that were kept in enclosures for more than a year. There has been increasing attention on the genus Acinetobacter due to its pathogenic properties and exceptional ability to develop drug resistance, especially the species Acinetobacter baumannii [33]. However, according to a study performed on black-headed gulls and songbirds, A. baumannii did not show a preference for avian hosts [33]. Since the genus Acinetobacter is not uncommon in soil, water and humans, including healthy individuals [34], the higher frequency and proximity of interactions with humans in a captive setting might have caused the enriched population of this genus.
4.2. The Effect of Urbanisation
One of the main objectives of this study was to identify any associations between urbanisation level and gut microbial shifts. Although no differences were identified in alpha diversity, the “Urban” population had a significantly higher abundance of Ligilactobacillus. Being a genus of lactic acid bacteria, Ligilactobacillus spp. play important roles in food digestion, absorption, and strengthening host resistance to pathogens, especially the species Ligilactobacillus salivarius [35]. An increase in their number could be a mitigating strategy against the toxic effects of polluted environments. This has been suggested in another study of the impact of heavy metal exposure on tree sparrows (Passer montanus), with an increase in Lactobacillus being one of the significant findings [17]. Lactobacillus and Ligilactobacillus are taxonomically very close and were classified, along with 24 other genera, as one genus until 2020 [36].
On the other hand, the bacterial family Staphylococcaceae from the phylum Bacillota, which is generally characterised by its digestion-aiding ability, had a higher abundance in “Suburban” groups than both “Urban” and “Captive” populations. This family is ubiquitous in bird microbiota, with a few exceptions of infectious species like Staphylococcus aureus [37]. Our finding is corroborated by studies on house sparrows in which the urban individuals exhibited lower Staphylococcaceae abundance [10,38].
The abundance of the genus Streptococcus was marginally higher in the “Suburban” population than in the “Urban” population. This might indicate the more diverse diet of the Herring Gulls inhabiting “Suburban” sites, as in a previous large-scale study, omnivorous wild birds were found to have a higher gut content of Streptococcus than granivorous species [39]. Observationally, active feeding by humans is more frequent in the “Urban” sites than in the “Suburban” sites. Although further research is needed for Herring Gulls, a study on anthropogenic feeding on yellow-legged gulls has highlighted bread as the most commonly fed item, which is made from grains [40].
4.3. Site-Specific Characteristics
We saw that the gut microbiome of birds from some of the study sites stood out from the others. First, “Bristol” is the only group with significant alpha diversity differences compared to other sites, as reflected by the Inverse Simpson index. This indicates a higher gut microbial diversity with an emphasis on species evenness. Considered as a highly urbanised site, “Bristol” inhabitants might be exposed to a higher diversity of pathogenic and toxin-related bacteria, resulting in a higher alpha diversity. The heightened diversity is accompanied by a significantly higher abundance of the phylum Bacillota and the family Ligilactobacillus, which could be a mitigating strategy, as discussed above. Although Bacillota are commonly regarded as beneficial bacteria, the domination of this phylum could be a result of the high-fat content in the diet, as recognised in human and rodent studies [41]. The overall characteristics of this site could possibly suggest a highly polluted environment with high-fat food made available by humans, which coincides with our observation of this study location. However, it is noteworthy that similar situations could also be observed in other urban sites, and therefore, this might not be the only reason behind the significant results. Likewise, several detected genera, including Bacillus and Clostridium, are spore-forming taxa that are widely distributed in the environment and commonly encountered as laboratory or reagent contaminants. Detection of these genera may represent transient individuals instead of part of the established stable gut microbiome community. As such, quantification of Bacillota may represent artificially inflated counts of Bacillus. While we did not detect significant amounts of either taxon in our negative controls, we cannot definitively distinguish environmental introduction from true colonisation.
Secondly, “Gloucester” samples showed significantly higher bacterial content of the family Lactobacillaceae and the genera Bacillus and Klebsiella. High abundance of Lactobacillaceae in more urbanised avian populations has also been recorded in two studies on house sparrows [10,38]. The genus Lactobacillus, Lactobacillaceae includes many probiotic species crucial for intestinal health, regulating the immune system and protecting against infections [42]. While it is generally believed that an abundance of probiotic-implicated taxa indicates healthy individuals, a strikingly higher abundance of Klebsiella was also found in this population. Although Klebsiella spp. are commonly isolated in the environment and also in the animal gastrointestinal tract, species like Klebsiella pneumoniae can lead to respiratory infections in birds [43]. This species is also highly dominant, making up 95% of the total Klebsiella spp. isolated from turkeys in a study [44]. Furthermore, the high presence of Klebsiella spp. in this gull population indicates the possibility of them acting as mobile reservoirs for the spread of Klebsiella pneumoniae. This could be of public health concern, as they are known to cause infections by the multidrug-resistant strains [45].
Thirdly, in the case of the “West Kirby” population, a significantly higher proportion of Fusobacteriota was identified. This result is in line with research on the gut microbiome of 13 migratory shorebird species [15] and one study on urban Herring Gull [15,46]. Fusobacteriota have been associated with a carnivorous and omnivorous diet, and since West Kirby is on the coast, a small town with access to human but also natural rocky and sandy shorelines, this may reflect that the Herring Gulls in West Kirby are likely to scavenge yet still have a wider range for foraging [46].
A higher proportion of Mycoplasma was detected in the “Liverpool” population. Interestingly, in a previous large-scale study of over 1000 birds of 50 species, including 16 Herring Gulls [47], this bacterial genus showed a high occurrence (~80–90%) [45], which suggests Mycoplasma species are a part of the common gull microbiome. Here we saw a much lower prevalence (30%). Among over 100 species of Mycoplasma, some are more pathogenic than others, like Mycoplasma gallisepticum and Mycoplasma synoviae, which cause avian mycoplasmosis, and most of the others are opportunistic pathogens [47]. While M. gallisepticum and M. synoviae were not detected in the previous study, it is suggested that there might be different Mycoplasma species unique to Laridae birds [47]. Overall, the high abundance of Mycoplasma in Liverpool Herring Gulls could suggest a possible outbreak of mycoplasmosis, although this may be detection of Mycoplasma species that are part of the normal microbiome. Therefore, further research is needed to parse the difference between a disease outbreak and a high prevalence of Mycoplasma colonisation as part of normal microbiome variation.
4.4. Limitations
In our study, we used opportunistic faecal sample collection as our sampling method, which was possible because of the abundance of Herring Gulls and their proximity to human residences. However, this method does not result in a high sample size relative to collection effort. This is especially apparent in the Steepholm Island, where only two samples were collected despite over seven hours of observation by four researchers. In addition, our sampling period overlapped with one of the hottest periods of the year, which could have led to dehydration and inactivity in birds. Our resulting sample size is smaller than ideal when compared to other wild avian gut microbial research. Because richness estimates can be influenced by sequencing depth, comparisons are restricted to overall trends rather than absolute differences between samples. Although long-read nanopore 16S sequencing can improve resolution, species-level classification remains sensitive to sequencing errors, primer bias, and reference database accuracy. All analysis were performed at the genus classification level or higher to avoid these uncertainties at the species level. Conclusions are based on higher-level taxonomic trends and relative differences between samples. Furthermore, our study was limited to a non-invasive sampling method, which could lead to a higher chance of contamination and a different representation of the host gut microbiome compared to intestinal mucosal samples [48].
5. Conclusions
Our study provides valuable insights into the possible influence of habitat location, types and settings on avian gut microbiome diversity and composition using Herring Gulls as the research subject. While the dominance of the Bacillota and Proteobacteria phyla remained consistent with other avian gut microbiome studies, differences could be found in some of the alpha diversity indices and microbial compositions between habitat types and sampling sites. Some of these differences are candidates for further investigation.
Firstly, the impact of captivity on the gulls might seem paradoxical, with a seemingly positive impact as a higher Shannon’s index could imply; however, there is a possible health concern regarding a higher abundance of Acinetobacter. Secondly, the higher abundance of Ligilactobacillus in the urban gulls might indicate an environment with more pollutants, such as heavy metals, raising concern about the possible adverse effect of pollution on wild birds. In addition, Bristol, one of the highly urban sites, displayed a higher abundance of Bacillota, which could be explained by the high-fat processed food that is readily available. Thirdly, the suburban gull populations could have a more diverse diet and a wider range for foraging and scavenging than the urban gulls, as implied by the higher abundance of Streptococcus in suburban gulls. Finally, four of our sampling sites, Bristol, West Kirby, Gloucester and Liverpool, displayed characteristic features identifying a high prevalence of Mycoplasma, which may suggest a possible outbreak of mycoplasmosis in Liverpool gulls.
Further investigation should focus on testing for particular pathogenic species, diet analysis, foraging patterns and conducting experiments focusing on possible factors, function and impact of certain microbes on the host’s health. Increased sample sizes would also add power to the analysis. A better understanding of how the avian gut microbiome interacts with the habitat, and the host’s health and behaviour, could aid conservation efforts in different aspects. It could be applicable in managing environmental disturbances, including pollution, controlling disease and possible zoonoses, and aiding health care for captive breeding gulls, especially vulnerable species like the relict gulls (Larus relictus) [49].
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Grimm N.B. Foster D. Groffman P. Grove J.M. Hopkinson C.S. Nadelhoffer K.J. Pataki D.E. Peters D.P.C. The Changing Landscape: Ecosystem Responses to Urbanization and Pollution Across Climatic and Societal Gradients Front. Ecol. Environ.2008626427210.1890/070147 · doi ↗
- 2Mc Kinney M.L. Effects of Urbanization on Species Richness: A Review of Plants and Animals Urban Ecosyst.20081116117610.1007/s 11252-007-0045-4 · doi ↗
- 3Mc Manus N. Holmes S.M. Louis E.E. Johnson S.E. Baden A.L. Amato K.R. The Gut Microbiome as an Indicator of Habitat Disturbance in a Critically Endangered Lemur BMC Ecol. Evol.20212122210.1186/s 12862-021-01945-z 34915861 PMC 8680155 · doi ↗ · pubmed ↗
- 4Neely W.J. Greenspan S.E. Stahl L.M. Heraghty S.D. Marshall V.M. Atkinson C.L. Becker C.G. Habitat Disturbance Linked with Host Microbiome Dispersion and Bd Dynamics in Temperate Amphibians Microb. Ecol.20228490191010.1007/s 00248-021-01897-334671826 · doi ↗ · pubmed ↗
- 5Peixoto R.S. Harkins D.M. Nelson K.E. Advances in Microbiome Research for Animal Health Annu. Rev. Anim. Biosci.2021928931110.1146/annurev-animal-091020-07590733317323 · doi ↗ · pubmed ↗
- 6Grond K. Sandercock B.K. Jumpponen A. Zeglin L.H. The Avian Gut Microbiota: Community, Physiology and Function in Wild Birds J. Avian Biol.201849 e 0178810.1111/jav.01788 · doi ↗
- 7Banks J.C. Cary S.C. Hogg I.D. The Phylogeography of Adelie Penguin Faecal Flora Environ. Microbiol.20091157758810.1111/j.1462-2920.2008.01816.x 19040454 · doi ↗ · pubmed ↗
- 8Benskin C.M.W.H. Rhodes G. Pickup R.W. Wilson K. Hartley I.R. Diversity and Temporal Stability of Bacterial Communities in a Model Passerine Bird, the Zebra Finch Mol. Ecol.2010195531554410.1111/j.1365-294X.2010.04892.x 21054607 · doi ↗ · pubmed ↗