The Genetic Landscape of Androgenetic Alopecia: Current Knowledge and Future Perspectives
Aditya K. Gupta, Daniel J. Dennis, Vasiliki Economopoulos, Vincent Piguet

TL;DR
Androgenetic hair loss is influenced by multiple genes and biological pathways, with genetic risk varying across populations and between men and women.
Contribution
The paper highlights recent genetic discoveries in androgenetic alopecia, emphasizing polygenic influences and the need for diverse and gender-specific studies.
Findings
AGA is a polygenic disorder involving multiple pathways like androgen signaling and follicle development.
Genetic risk varies across populations, with limited transferability of findings from European cohorts to other ancestries.
Emerging data suggest distinct genetic risk factors in women compared to men.
Abstract
Androgenetic hair loss is the most common cause of gradual hair thinning in adults. For many years, this form of hair loss was thought to be driven mainly by male hormones and inherited in a simple way. Recent genetic research has shown that hair loss is influenced by many genes that affect how hair follicles grow, survive, and respond to hormones. These genes act through several biological pathways, leading to progressive thinning of hair follicles over time. Genetic risk also differs between populations, meaning that results from one ancestry group may not apply equally to others. In women, the genetic factors involved in hair loss appear partly different from those in men, and more research focused on women is still needed. Studies are also beginning to show that a person’s genetic makeup may influence how well certain hair loss treatments work or whether side effects might occur.…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
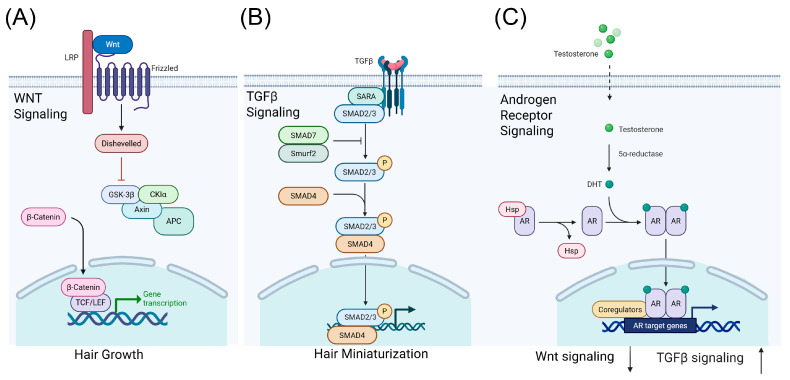 Figure 1
Figure 1| Gene/Locus | SNP or | Population | Risk |
|---|---|---|---|
| rs6152 | European mixed ancestry male | Increased risk | |
| rs12558842 | European mixed ancestry male population; Mixed ethnicity mixed sex; German male population | Increased risk | |
| Xq12 locus [ | rs1041668 | European mixed ancestry male population | Increased risk |
| 20p11 locus [ | rs1160312 | European mixed ancestry male population | Increased risk |
| rs6113491 | European mixed ancestry male population | Increased risk | |
| rs7349332 | European mixed ancestry male population | Increased risk | |
| rs7648585 | European mixed ancestry male population | Increased risk | |
| rs929626 | European mixed ancestry male population | Reduced risk [ | |
| rs1081073 | European mixed ancestry male population | Increased risk [ | |
| rs9668810 | European mixed ancestry male population | Increased risk | |
| rs7975017 | European mixed ancestry male population | Increased risk | |
| rs2073963 | German male population; European mixed ancestry mixed sex population | Increased risk | |
| rs12565727 | European mixed ancestry male population; European mixed ancestry mixed sex population | Reduced risk [ | |
| rs6945541 | European mixed ancestry mixed sex population | Increased risk | |
| rs6047844 | European mixed ancestry mixed sex population, | Increased risk | |
| rs13283456 | Mixed ethnicity and mixed sex | Increased risk | |
| rs523349 | Mixed ethnicity and mixed sex | Increased risk | |
| rs1800012 | Mixed ethnicity and mixed sex | Increased risk | |
| rs4343 | Mixed ethnicity and mixed sex | Increased risk | |
| rs10782665 | Mixed ethnicity and mixed sex | Increased risk | |
| rs533116 | Mixed male and female | Increased risk | |
| rs545659 | Mixed ethnicity and mixed sex | Increased risk | |
| rs12724719 | Mixed ethnicity and mixed sex | Increased risk | |
| Not known [ | rs11010734 | Korean male population | Increased risk |
| rs2420640 | Korean male population | Increased risk | |
| 2q31.1 locus [ | rs13405699 | Han Chinese male population | Increased risk |
| rs982804 | European mixed ancestry male population | Increased risk | |
| rs12203592 | European mixed ancestry male population | Increased risk | |
| rs145945174 | European mixed ancestry male population | Increased risk [ | |
| rs116494345 | African male population | Increased risk [ | |
| rs143451223 | African male population | Increased risk | |
| rs2163085 | Korean female population | Increased risk | |
| rs4793158 | Korean female population | Increased risk |
| Author | Select Genes/Loci | Study Type | Sample Size (Cases/Total N) | Strength of Evidence |
|---|---|---|---|---|
| Ambra et al. 2025 [ | Xq12 locus, 20p11 locus | Genetic association study | 104 cases/ | Moderate—single study with a relatively small sample, associations adjusted for confounders, but no replication cohort or meta-analysis included |
| Brockschmidt et al. 2011 [ | GWAS | 581 cases/ | Moderate to strong—replicated in independent sample; supported by fine-mapping, family-based TDT analysis, and tissue expression studies; effect sizes modest | |
| Francès et al. 2024 [ | Candidate SNP association study | 26,607/ | Low to moderate—Large sample size improves statistical power, However, restricted to predefined candidate SNPs. Associations reported at nominal significance thresholds ( | |
| Heilmann et al. 2013 [ | Replication of meta-analysis | 2759/5420 N (plus previous meta-analysis) | Strong—genome-wide significant loci confirmed, multi-cohort replication, robust QC and statistical methods, supported by expression analysis in human hair follicles | |
| Heilmann-Heimbach et al. 2017 [ | GWAS | 10,846/ | Very strong—high-quality genetic evidence. Genome-wide significance threshold applied ( | |
| Henne et al. 2023 [ | Exome wide association | 72,469/ | Strong—very large, population-based exome sequencing study; combines single-variant and gene-based tests; confirms known genes and identifies novel rare variant associations; results are further integrated with PRS for risk modeling | |
| Janivara et al. 2025 [ | GWAS | 2136/ | Moderate—limited by moderate sample size for detecting genome-wide significance and by reliance on self-reported baldness | |
| Kim et al. 2022 [ | GWAS | 275/421 N | Low to moderate—Single-center, hospital-based cohort, modest sample size with limited statistical power, GWAS findings reach suggestive significance rather than conventional genome-wide significance, replication signals largely nominal ( | |
| Lee et al. 2024 [ | GWAS | 545/1004 N | Moderate—Single-cohort GWAS with modest sample size, includes replication of known loci, limited power relative to large meta-analyses | |
| Li et al. 2012 [ | Meta-analysis of GWAS | 3891/ | Strong—Large-scale GWAS meta-analysis across multiple cohorts, genome-wide significant loci identified ( | |
| Li et al. 2024 [ | 2q31.1 | Candidate SNP replication | 499/1988 N | Moderate—Well-powered replication of known GWAS loci, but only 1 SNP reached significance after multiple testing, limited by relatively small sample size |
| Marcińska et al. 2015 [ | Candidate SNP association study | 476/605 N | Moderate—validated in independent test set, moderate sample size | |
| Zhuo et al. 2012 [ | Meta-analysis | 2074/3189 N | Moderate—synthesizes multiple studies, limited by small number of included studies ( |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsHair Growth and Disorders · Facial Rejuvenation and Surgery Techniques · Lipid metabolism and disorders
1. Introduction
Androgenetic alopecia (AGA), commonly known as male-pattern or female-pattern hair loss, is the most prevalent form of progressive hair thinning in adults. Epidemiological studies estimate that nearly 70% of Caucasian men will experience some degree of AGA by age 70 years, and nearly 50% of women [1,2,3]. Men typically have an earlier onset of hair loss than women [4]. The condition is characterized histologically by the miniaturization of terminal hair follicles and a shortening of the anagen (growth) phase [5,6], driven by androgen signaling in genetically susceptible hair follicles [7]. Genetically, AGA is among the most heritable dermatological traits: twin and familial studies have suggested heritability estimates up to ~80%, pointing to a strong polygenic architecture [8,9].
However, early candidate-gene association studies, most notably involving the androgen receptor (AR) locus on the X chromosome and the 20p11 region, accounted for only a fraction of the inherited risk [10], highlighting the need for more comprehensive genome-wide efforts.
In recent years, large-scale genome-wide association studies (GWAS) have dramatically expanded our understanding of AGA’s genetic architecture. A large GWAS identified 71 AGA risk loci, and highlighted genes across multiple biological pathways, such as WNT signaling [11].
Similarly, a key meta-analysis implicated biological pathways such as melatonin signaling and adipogenesis [12]. Despite this progress, much of the heritable risk for AGA remains unexplained, and the discovery of risk alleles has been uneven across populations. For instance, studies in populations of non-European ancestries have validated some established loci while also identifying novel variants [13,14,15], underscoring the importance of ancestry-specific studies. Given these advances, there is a critical need to translate genetic findings into biological mechanisms, clinical risk prediction, and ultimately therapeutic strategies. Functional characterization of GWAS loci, integration with multi-omics and single-cell data, and pharmacogenetic insights are increasingly informing our understanding of how genetic variation shapes both disease susceptibility and treatment response. In this review, we synthesize the current literature on AGA genetics, with a focus on genome-wide risk loci, mechanistic pathways, sex and ancestry differences, and the emerging clinical implications for pharmacogenetics.
2. Genetic Architecture of AGA
2.1. Genome-Wide Association Studies
Over the past decade, genome-wide association studies have defined AGA as a highly polygenic trait with dozens of loci of small to moderate effect. Early studies built on classic candidate gene findings at the androgen receptor (AR) locus and the 20p11 region, but expanded the architecture substantially [16,17].
A landmark GWAS in over 70,000 men of European ancestry identified 71 independent susceptibility loci, explaining approximately 38% of the single nucleotide polymorphism (SNP)-based risk for AGA, and implicating genes across biologically coherent pathways including WNT signaling, androgen metabolism, apoptosis, and morphogenesis [11]. This built on an earlier meta-analysis that had expanded candidate loci to implicate genes such as FGF5, IRF4, DKK2, and pathways including melatonin signaling and adipogenesis [12]. Because many participants in these studies were aged 50 years and older, it is plausible that some of the non-androgenic loci (e.g., WNT-pathway) reflect a mixture of classical AGA and age-related (senescent) hair thinning, rather than AGA alone [18].
While functional analysis has not been performed on most SNPs, more recent post-GWAS approaches, including transcriptome-wide association studies (TWAS) have begun to identify more functional gene-level candidates [19]. The study identified several novel genes, including CD59 (cell survival and apoptosis), ZDHHC5 (membrane localization), and ZIC2 (follicle morphogenesis). While most of the work on identifying SNPs has been in individuals with European ancestry, recent GWAS have used subjects with non-European ancestry (discussed in under Ancestry considerations). Although genetic studies of AGA have largely focused on common variants identified by genome-wide association studies, analysis of recent large-scale exome sequencing data from the UK Biobank suggests that rare genetic variants play a relatively small role in AGA risk, with associations limited to few (5) genes, two of which (EDA2R and WNT10A) are already implicated by GWAS, rather than evidence for rare, high-impact mutations [20].
2.2. Key Biological Pathways
Although GWAS loci are dispersed across the genome, functional annotation and follow-up studies repeatedly implicate a limited number of coherent biological pathways. Prominent among these are androgen metabolism/AR-mediated signaling [21,22,23], WNT/β-catenin signaling (for example WNT10A and its regulatory partners) [24], pathways controlling hair-follicle morphogenesis and stem/progenitor cell function [25], and pathways related to apoptosis, extracellular matrix remodeling, and energy metabolism (Table 1) [25,26]. Expression studies in human scalp follicles from AGA patients reveal altered regulation of extracellular matrix and progenitor cell genes, suppression of canonical WNT/β-catenin signaling, increases in TGF-β signaling (which can lead to hair miniaturization through premature transition to the catagen phase) [27], and dysregulated structural and metabolic pathways in miniaturizing follicles (Figure 1) [25,26]. Together, these findings support a model in which genetic variation influences follicle regeneration and susceptibility to miniaturization in androgenetic alopecia [21,22].
2.3. Sex Differences
Sex modifies both epidemiology and genetic architecture in patterned hair loss [34]. Classic candidate-gene work and later genomic studies show a major contribution from variants at the AR locus on the X chromosome, explaining part of the strong male bias in many cohorts [2]. However, when known male AGA risk loci have been tested in women with female-pattern hair loss (FPHL), results have been largely negative or inconclusive [32,35]. This may reflect not only true biological differences between male- and female-patterned hair loss, but also substantial phenotypic heterogeneity and diagnostic ambiguity within FPHL cohorts. In clinical practice and epidemiologic studies, diffuse hair thinning in women is frequently classified as FPHL despite the possibility that multiple biological processes, such as chronic telogen effluvium, age-related (senescent) alopecia, or mixed phenotypes, may contribute to the observed pattern [36,37]. Such phenotypic misclassification is likely to dilute genetic signals and reduce power to detect reproducible susceptibility loci.
Early attempts to replicate male AGA loci in women found no significant association at 20p11 and only a nominal AR/EDA2R signal, with subsequent analysis of additional male loci failing to replicate in FPHL cohorts [35,38]. Although these findings raised the question of whether FPHL might instead reflect senescence-related hair thinning, current evidence suggests that age-related changes alone cannot fully account for the phenotype [39]. As in male cohorts enriched for older individuals, where classical AGA may coexist with senescent alopecia, genetic studies in women are similarly vulnerable to phenotypic admixture. Diffuse thinning in women may reflect a mixture of androgen-dependent FPHL, age-related hair thinning, and non-androgenetic processes, which can obscure true genetic associations and contribute to inconsistent replication across studies [36,39].
More recent work supports the presence of female-specific contributions [32]. In a sex-stratified genome-wide association study of a Korean population, several loci reached significance exclusively in women, such as rs2163085 (FZD1) and rs4793158 (GJC1) [32]. These findings provide some of the first genome-wide evidence that the genetic determinants of FPHL are at least partially distinct from those underlying male-pattern hair loss. However, the current literature remains limited by small sample sizes, heterogeneous case definitions, and inconsistent phenotyping, making it difficult to disentangle true sex-specific genetic architecture from methodological confounding. Extrapolating risk models from male-derived GWAS to women due to a lack in female-only studies is likely insufficient, highlighting the need for well-powered female-specific GWAS.
2.4. Ancestry Considerations
Most published discovery GWAS have used European-ancestry cohorts, which has two consequences: (1) many discovered loci have been optimized to European linkage disequilibrium (LD) patterns and allele frequencies, meaning that the lead SNPs identified in these studies are the variants that best tag the underlying causal variants in European populations but may not tag them well in other ancestries, and (2) polygenic risk scores trained in Europeans attenuate when applied to non-European populations. In European populations, AR variants have been linked not only to AGA risk but also to earlier age of onset [22], providing a direct connection between genetic architecture and observed population-level clinical features. These issues have started to be addressed by a recent shift in the field toward ancestry-diverse studies.
Independent studies in East Asian and African populations have both replicated several canonical loci, such as 20p11 and 2q31.1, and identified additional, population-specific signals [13,15,32,40], underscoring the need for broader ancestral representation across studies of varying design and statistical power (Table 2).
Recent work provides some of the first broad replication and pilot genome-wide evidence for AGA risk in Asian populations. A Korean study confirmed that SNPs in the 20p11 region are associated with AGA, consistent with findings from European cohorts, but variants in AR and EDA2R were not associated in Korean individuals [13]. Importantly, the study identified two SNPs, rs11010734 and rs2420640, found at 10p11.21 and near PANK1-KIF20B, respectively, that were uniquely associated with AGA risk in Korean populations [13]. Much like Korean populations, the 20p11 region has been previously associated with AGA risk in Chinese populations [41]. In addition, Chinese populations had shown an association with the 2q31.1 SNP rs13405699 and several other genes, including WNT10A (rs7349332), that had previously been reported in populations with European ancestry [14]. Population-level observations suggest that AGA tends to present with later onset and more diffuse hair thinning in East Asian men compared with Europeans [42], although the specific contribution of these SNPs to these clinical differences remains to be formally established.
Very recent work in African men by Janivara et al. found that European-derived polygenic risk scores for AGA performed poorly in African men, showing minimal predictive ability [15]. The authors identified several loci associated with AGA, including 36 associations that appear unique to African populations, including rs116494345 at 1p13.2 and rs143451223 at 1q41 [15]. The strongest signal was at 1p13.2, where the lead SNP rs116494345 is monomorphic in European and Asian populations, representing a truly African-specific association. Most AGA-associated variants in this study were autosomal, and unlike findings in European cohorts, the X chromosome did not show strong associations with AGA in African men. Epidemiological studies indicate that African men generally exhibit lower overall prevalence of AGA [43], consistent with the possibility of population-specific genetic effects.
Taken together, these findings highlight that the genetic architecture of AGA is partially population-specific. This has important implications, as polygenic risk scores or therapeutic predictions derived from European cohorts may not generalize to other ancestries, and novel, ancestry-specific loci may reveal new biological pathways or targets for intervention.
3. Genetic Associations and Biological Mechanisms
3.1. Functional Annotation of GWAS Loci
GWAS have repeatedly implicated the AR locus and an androgen-independent signal at 20p11 as major AGA susceptibility regions [11]. Translating locus associations to genes requires ancestry-aware fine-mapping, integration with expression quantitative trait loci (eQTL) and chromatin annotations, and colocalization to prioritize candidate causal genes and regulatory elements [45,46]. Such functional annotations have emphasized that many lead SNPs are tag variants and that differing linkage disequilibrium and allele frequencies between ancestries change which variants best tag a locus [47].
3.2. Insights from Single-Cell and Multi-Omics Studies
Single-cell and spatial transcriptomic work on human scalp and follicles is beginning to map cell types and regulatory states relevant to hair cycling [48,49,50]. Single-cell atlases of human scalp hair follicles provide lineage maps that can help localize GWAS-nominated genes to specific follicular cell compartments.
While AGA-specific single-cell studies remain limited, recent work from Ober-Reynolds et al. have utilized this approach to link AGA GWAS loci with regulatory elements in scalp tissue [51]. Notably, they observed strong enrichment of AGA-associated SNPs within open chromatin regions in dermal papilla cells, implicating this mesenchymal population as a key mediator of genetic risk [51]. They further predicted that WNT10A SNP rs72966077 was functionally important in AGA, providing mechanistic insight into how at least some risk loci may influence hair-follicle biology.
Single-cell studies of human scalp follicles further resolve epithelial and progenitor populations, enabling future integration of transcriptomic data to determine cell-type-specific mechanisms in AGA [45,46]. Together, these findings suggest that single-cell and chromatin mapping can link GWAS loci to candidate genes and regulatory networks, generating hypotheses for how biological pathways contribute to AGA.
3.3. Clinical Significance of Biological Mechanisms
The strongest translationally actionable mechanism for AGA remains androgen signaling, with AR and local dihydrotestosterone (DHT) biology rationalizing 5-alpha reductase inhibitors and AR-targeted approaches for many patients [45]. However, loci such as 20p11 and multiple other autosomal GWAS hits implicate androgen-independent pathways (e.g., WNT/TGF-β) that likely contribute to variable treatment response and offer alternative therapeutic targets [11,12,44]. The WNT signaling pathway is important for regulating hair follicle regeneration [52]. A WNT10A polymorphism (rs7349332) is strongly associated with AGA and may result in lower WNT10A expression [14,24]. Topical tretinoin may be a potential treatment to target the WNT pathway, as improved hair growth was observed in a recent clinical study [53].
4. Pharmacogenetics and Clinical Implications
4.1. Finasteride and Dutasteride
Finasteride and dutasteride reduce DHT by inhibiting type II and type I/II 5-α-reductase, respectively, and are the most effective systemic therapies for AGA in men [54]. Although genetic predictors of treatment response have been proposed, no validated pharmacogenetic markers currently guide finasteride or dutasteride use in AGA.
Studies of SRD5A2 variants have shown functional effects in prostate disorders but have not demonstrated consistent associations with clinical response in AGA [40,55]. Overall, pharmacogenetic stratification for 5-α-reductase inhibitors remains investigational, and therapeutic decisions rely on clinical factors rather than genotype. Larger studies are still needed before genetic testing can predict finasteride and dutasteride outcomes.
In terms of treatment, hormonal therapies such as finasteride and dutasteride are effective in men with AGA [56], but clinical trials in women show variable efficacy, reflecting both hormonal differences and the distinct genetic architecture of FPHL [57]. While functional analysis has not been performed on most SNPs, a more recent transcriptome-wide association study (TWAS) was performed to identify more functional gene-level candidates [19]. The study identified several novel genes, including CD59 (cell survival and apoptosis), ZDHHC5 (membrane localization), and ZIC2 (follicle morphogenesis). While most of the work on identifying SNPs has been in individuals with European ancestry, recent GWAS have used subjects with non-European ancestry (discussed in under Ancestry considerations).
4.2. Minoxidil
Topical minoxidil requires activation to minoxidil sulfate by sulfotransferase SULT1A1 [58]. Early work demonstrated that low SULT1A1 activity in plucked hair follicle samples predicts poor response to topical minoxidil [59]. However, a recent study of AGA and oral minoxidil found low SULT1A1 correlated with better response [60], indicating that the relationship between SULT1A1 and minoxidil may depend on the route of administration or other modifiers. Recent genetic data from a 26-SNP panel study found that SULT1A1 variant rs1042028 was a predictor of poor minoxidil response, suggesting a role for inherited variation in drug responsiveness [61]. Overall, while SULT1A1 remains a good candidate pharmacogenetic marker for minoxidil response, further large-scale controlled studies are required before routine clinical implementation.
4.3. Genetics and Adverse Effects
Reports of persistent sexual or neuropsychiatric symptoms after finasteride remain rare and causality is unconfirmed. Presently, adverse events have not been robustly linked to reproducible genetic polymorphisms across populations. However, early pharmacogenetic studies reported that repeat-length polymorphisms in the AR gene, rs4045402 and rs3138869, were more frequent among AGA patients reporting persistent side-effects after finasteride (post-finasteride syndrome) compared with controls [62,63]. For minoxidil, no convincing pharmacogenetic safety markers have been described to date. CYP3A5 SNPs are biologically plausible candidates for altered finasteride metabolism, as homozygous carriers of rs776746 have lower finasteride clearance in the liver [56]. One small pilot genotyping study did include the CYP3A5 variant rs776746 (a candidate for drug metabolism), but it did not demonstrate clear associations with adverse effects or drug clearance in that cohort [64].
4.4. Other Therapeutic Approaches and Pharmacogenetic Considerations
Several additional therapies are used or are under investigation for androgenetic alopecia, although pharmacogenetic evidence for these approaches is limited. Topical anti-androgens (e.g., spironolactone and flutamide) have shown modest efficacy, particularly in women, but no reproducible genetic predictors of response or safety have been identified [65,66,67].
Low-level light therapy and platelet-rich plasma have demonstrated variable clinical benefit in controlled studies; however, inter-individual differences in response have not been linked to specific genetic variants [68,69]. Other emerging approaches, including botulinum toxin, and agents targeting WNT signaling, remain investigational, and pharmacogenetic data are currently lacking [52,70]. Recent evidence suggests that tretinoin may represent a novel therapeutic approach [53]. Experimental studies show that topical tretinoin promotes hair follicle stem cell activation, accelerates the transition from telogen to anagen, and stimulates hair growth via Wnt/β-catenin pathways [53]. Early clinical data indicate that tretinoin can improve hair counts and density in AGA patients [53]. However, pharmacogenetic predictors of response to RA therapy have not yet been identified.
Overall, outside of androgen suppression and minoxidil activation, genetic modifiers of treatment response for alternative AGA therapies remain largely unexplored, highlighting an unmet need for genotype-stratified clinical studies.
5. Future Directions
5.1. Research Needs
Moving GWAS findings toward clinical application will require ancestry-aware fine-mapping and functional follow-up to nominate causal variants and cell types, because LD differences mean that GWAS lead variants often do not mark the same causal allele across ancestries. Large, diverse cohorts plus integrated fine-mapping using chromatin annotations and eQTL/colocalization will reduce false leads from tag SNPs and LD differences across populations. Single-cell and spatial multi-omic atlases of human scalp follicles must be expanded and linked to genotypes so that noncoding GWAS signals can be assigned to precise regulatory elements and cell types.
Recent work demonstrating the power of integrated single-cell chromatin + transcriptome maps in skin and hair tissues exemplifies this approach [51]. Although spatial ATAC-seq and related spatial epigenomic methods are still emerging, applying them to scalp tissue would be especially informative for AGA, where many genetic associations are noncoding and likely act through regulatory elements that require in-tissue context to interpret. In parallel, the field needs well-powered pharmacogenetic studies (genotyped clinical cohorts) that prospectively measure treatment response and adverse events for finasteride/dutasteride and minoxidil, including mechanistic biomarkers (e.g., follicular SULT1A1 activity for minoxidil activation). Small trials and biomarker studies are encouraging but underpowered; larger, standardized collections are essential.
Beyond medical therapy, genetic influences on surgical interventions such as hair transplantation remain poorly characterized. Although the principle of donor dominance explains why transplanted occipital follicles, genetically resistant to androgen-mediated miniaturization, generally maintain growth [71], no specific genetic variants or polygenic risk models are currently available to predict graft survival, cosmetic outcome, or long-term success in individual patients. At present, surgical technique and ongoing progression of native hair loss remain the primary determinants of clinical outcome [71], with genetic predictors representing a future research direction.
An additional priority is improved phenotypic definition in genetic studies of patterned hair loss, particularly in women. Standardized diagnostic criteria that distinguish FPHL from chronic telogen effluvium, senescent alopecia, and mixed phenotypes will be essential to reduce misclassification and enhance statistical power. Incorporating clinical patterning, trichoscopic features, hormonal context, and age stratification into GWAS design may clarify true sex-specific genetic architecture and improve reproducibility of susceptibility loci.
5.2. Prospective Studies
Prospective, genotype-stratified clinical trials should be prioritized. Examples of high-value designs include (a) randomized trials of topical/oral minoxidil with pre-treatment measurement of follicular SULT1A1 activity or SULT1A1 genotype to validate predictive value, and (b) trials testing 5-α-reductase inhibitors where SRD5A and AR variants are prespecified moderators of efficacy and safety.
Pilot trials that used SULT1A1 boosters [72] or measured enzyme activity [59,73] show proof of principle but need replication in larger, multi-center cohorts with standardized endpoints. Prospective pharmaco-safety registries that link longitudinal adverse-event reporting with genotype data are also needed to evaluate putative rare, persistent side effects of finasteride and to search for genetic risk markers in an unbiased way. Such registries should use harmonized case definitions and include neuropsychiatric and sexual health outcomes.
5.3. Therapeutic Potential
Integrative genomics and multi-omics will reveal new druggable pathways beyond androgen signaling, for example, WNT-modulation, TGF-β axis interventions, and paracrine factors from dermal papilla, which are now being explored in preclinical and early clinical work [74,75]. Reviews of recent drug development efforts highlight small molecules, biologics, and cell-based approaches that could be prioritized by genetics and cell-type evidence.
Near-term translational opportunities include using SULT1A1 activity/genotype to personalize minoxidil prescribing [76] and testing topical finasteride/minoxidil combinations or local AR modulators to minimize systemic exposure [77,78,79]. In the longer term, genotype-informed selection of patients for WNT or stem-cell–directed therapies could improve response and reduce off-target effects; however, rigorous causal validation is required before clinical deployment.
6. Conclusions
An increased understanding of the genetics underlying androgenetic alopecia has shifted the view of AGA from the classic, highly heritable, androgen-driven, monogenic trait to a polygenic condition with numerous risk loci converging on multiple biological pathways, including androgen signaling, WNT/TGF-β pathways, apoptosis, and follicular morphogenesis.
Emerging data also indicate that the genetic architecture of female-pattern hair loss differs from that of men, yet female-focused GWAS remain limited. Sex and ancestry influence both susceptibility and the predictive value of genetic variants, underscoring the need for ancestrally diverse, sex-specific studies.
Although GWAS have identified many associations, much of the heritable risk remains unexplained, and functional characterization of causal variants is limited. Emerging single-cell and spatial multi-omic approaches offer the potential to map noncoding variants to specific follicular cell types and regulatory elements. Pharmacogenetic evidence suggests that variation in genes such as SULT1A1, CYP3A5, AR, and SRD5A2 may affect treatment response and safety, but validated markers for clinical use are not yet available.
Future research should prioritize ancestry-aware fine-mapping, integrative functional studies, and prospective, genotype-stratified clinical trials. Near-term applications include using SULT1A1 activity to personalize minoxidil therapy and refining local modulation of androgen signaling, while longer-term opportunities may involve targeting WNT/TGF-β pathways and stem/progenitor cell populations. Integrating genomic, transcriptomic, and pharmacogenetic data will be essential to realize precision, mechanism-driven management of AGA.
7. Methods
A narrative literature review was conducted using PubMed. PubMed searches combined terms for androgenetic alopecia (“androgenetic alopecia,” “male pattern hair loss,” “female pattern hair loss”) with genetic and pharmacogenetic keywords (“genetics,” “GWAS,” “exome sequencing,” “rare variants,” “polygenic risk score”) and clinical outcomes (“pharmacogenetics,” “hair transplantation,” “clinical outcomes”). Searches included publications from 2000 to 2025, limited to studies in humans and published in English. Additional references were identified from bibliographies of relevant articles. No formal systematic review or meta-analysis protocol was followed.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Devjani S. Ezemma O. Kelley K.J. Stratton E. Senna M. Androgenetic Alopecia: Therapy Update Drugs 20238370171510.1007/s 40265-023-01880-x 37166619 PMC 10173235 · doi ↗ · pubmed ↗
- 2Lolli F. Pallotti F. Rossi A. Fortuna M.C. Caro G. Lenzi A. Sansone A. Lombardo F. Androgenetic Alopecia: A Review Endocrine 20175791710.1007/s 12020-017-1280-y 28349362 · doi ↗ · pubmed ↗
- 3Martinez-Jacobo L. Villarreal-Villarreal C.D. Ortiz-López R. Ocampo-Candiani J. Rojas-Martínez A. Genetic and Molecular Aspects of Androgenetic Alopecia Indian J. Dermatol. Venereol. Leprol.20188426326810.4103/ijdvl.IJDVL_262_1729595184 · doi ↗ · pubmed ↗
- 4Gupta A.K. Wang T. Economopoulos V. Epidemiological Landscape of Androgenetic Alopecia in the US: An All of Us Cross-Sectional Study P Lo S ONE 202520 e 031904010.1371/journal.pone.031904040014580 PMC 11867384 · doi ↗ · pubmed ↗
- 5Paus R. Cotsarelis G. The Biology of Hair Follicles N. Engl. J. Med.199934149149710.1056/NEJM 19990812341070610441606 · doi ↗ · pubmed ↗
- 6Pierard-Franchimont C. Piérard G.E. Teloptosis, a Turning Point in Hair Shedding Biorhythms Dermatology 200120311511710.1159/00005172311586007 · doi ↗ · pubmed ↗
- 7Inui S. Itami S. Molecular Basis of Androgenetic Alopecia: From Androgen to Paracrine Mediators through Dermal Papilla J. Dermatol. Sci.2011611610.1016/j.jdermsci.2010.10.01521167691 · doi ↗ · pubmed ↗
- 8Nyholt D.R. Gillespie N.A. Heath A.C. Martin N.G. Genetic Basis of Male Pattern Baldness J. Investig. Dermatol.20031211561156410.1111/j.1523-1747.2003.12615.x 14675213 · doi ↗ · pubmed ↗