Mitochondrial genome of Acheilognathus striatus characterisation and phylogenetic analysis
Ruixin Yang, Jinhui Yu, Yongtao Tang, Chuanjiang Zhou

TL;DR
This study analyzed the mitochondrial genome of Acheilognathus striatus to better understand its evolutionary relationships with other fish species.
Contribution
The study provides the first complete mitochondrial genome sequence of Acheilognathus striatus and its phylogenetic placement.
Findings
The mitogenome is 16,692 bp long and includes standard mitochondrial genes.
A. striatus is phylogenetically closely related to R. shitaiensis.
The mitogenome shows AT skewness and anti-G bias.
Abstract
In this study, the taxonomic position of Acheilognathus striatus was clarified through mitogenome analysis. The circular mitogenome is 16,692 bp long and comprises 13 protein-coding genes, two ribosomal RNA genes, 22 transfer RNA genes, and one non-coding D-loop region. The mitogenome exhibits AT skewness and anti-G bias. Phylogenetic trees were constructed using 22 Acheilognathinae species, with Gobioninae and Leuciscinae used as outgroups. The phylogenetic tree revealed that A. striatus formed a sister group with R. shitaiensis. This study contributes to a better understanding of the mitogenome characteristics and evolutionary relationships among Acheilognathinae species.
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
 Figure 1
Figure 1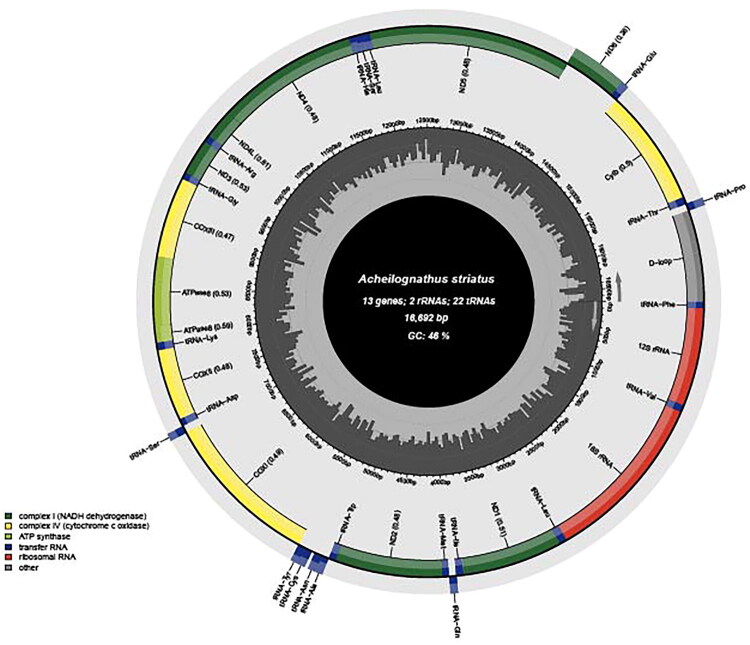 Figure 2
Figure 2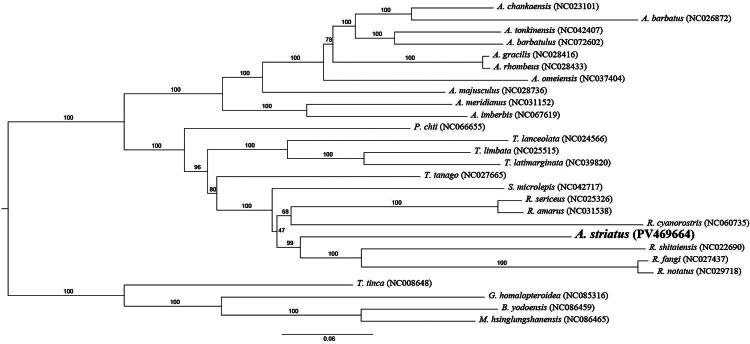 Figure 3
Figure 3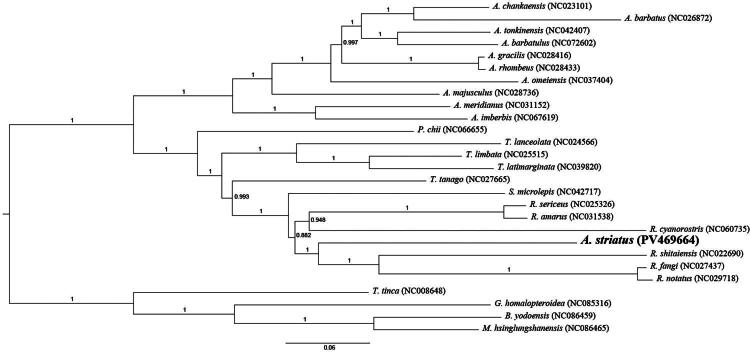 Figure 4
Figure 4- —National Natural Science Foundation of China10.13039/501100001809
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsGenomics and Phylogenetic Studies · Diatoms and Algae Research · Ichthyology and Marine Biology
Introduction
Acheilognathus striatus (Yang et al. 2010) is a freshwater fish species native to the Yangtze River, characterized by long barbels and a prominent black stripe extending from the body to the caudal peduncle. The last unbranched rays of both the dorsal and anal fins are thicker than the first branched rays. A. striatus belongs to the subfamily Acheilognathinae of the family Cyprinidae (Yang et al. 2010). Due to its ornamental value, A. striatus has been heavily exploited in its natural habitats, resulting in significant population decline. However, its phylogenetic placement has not yet been clearly resolved at the molecular level.
Mitogenome data of Acheilognathinae, Gobioninae, and Leuciscinae were retrieved from the NCBI database. Biwia yodoensis, Microphysogobio hsinglungshanensis, Gobiobotia homalopteroidea, and Tinca tinca were selected as outgroups. Thirteen PCGs were concatenated, and Bayesian inference (Ronquist and Huelsenbeck 2003) and maximum likelihood analyses (Stamatakis 2006) were performed based on optimal nucleotide substitution models and best partitioning schemes (Wang et al. 2025). This study provides valuable insights into the phylogeny and evolutionary origins of Acheilognathinae species.
Materials and methods
Sample collection, preservation, and DNA extraction
Samples of A. striatus (Figure 1) were collected alive and anesthetized prior to processing. The specimens were obtained from Huangshan City, Anhui Province (29°43′2.1ʺN, 118°19′56.9ʺE). Voucher specimens were deposited at the College of Life Sciences, Henan Normal University (voucher no. 23112101; contact: Chuanjiang Zhou, [email protected]). Genomic DNA was extracted using the phenol-chloroform method (Gautam 2022). High-throughput sequencing at 30× coverage was performed by PersonalBio (Nanjing) Biotechnology Co., Ltd.
Photograph of a female A. striatus specimen was collected from huangshan city, Anhui province (29°43′2.1ʺN, 118°19′56.9ʺE). The image was provided by jinhui Yu.
Mitogenome annotation, visualization, and comparative analysis
The mitochondrial genome of A. striatus was assembled and annotated from high-throughput sequencing data using GetOrganelle (Jin et al. 2020). Annotation was performed based on the codon usage of teleost fish, followed by verification of the circular conformation of the mitogenome. Mitofish was employed to analyze genomic structure, including composition, position, and gene length (Zhu et al. 2023) (Figure 2). A complete gene map of the mitogenome was constructed.
Gene map of the circular mitogenome of A. striatus generated with mitofish. The outer ring represents the light strand, and the inner ring represents the heavy strand. The GC content, gene composition, and length are illustrated in the center.
PhyloSuite v1.2.3 was used for amino acid translation, base composition analysis, and Relative Synonymous Codon Usage (RSCU) calculation (Zhang et al. 2020; Xiang et al. 2023). CodonW 1.4.2 was applied to compute the effective number of codons, codon adaptation index, codon bias index, and GC3 content (Puigbò et al. 2008) (Table S1).
Phylogenetic analyses
To determine the phylogenetic position of A. striatus, 26 species from Acheilognathinae, Gobioninae, and Leuciscinae were selected from the NCBI database. B. yodoensis, M. hsinglungshanensis, G. homalopteroidea, and T. tinca were used as outgroups. Phylogenetic trees were constructed using Bayesian inference and maximum likelihood analyses in PhyloSuite v1.2.3. PartitionFinder identified the best partitioning schemes and substitution models according to the Bayesian information criterion (Penny et al. 2007). ModelFinder determined the optimal model based on the Akaike information criterion (Kalyaanamoorthy et al. 2017).
Data resources
Complete mitogenome sequences of the 26 species were downloaded from the NCBI GenBank database (Table S2). The sequencing data for A. striatus were deposited in the National Center for Biotechnology Information under accession number PV469664.
Results
Genome structure and characteristics
The mitogenome of Acheilognathus striatus was found to be 16,692 bp in length, comprising 13 protein-coding genes (PCGs), 22 tRNAs, two rRNAs, and one D-loop control region, like for most other fish species. Except for eight tRNAs and the NAD6 gene, located on the light strand, the remaining 28 fragments are encoded on the heavy strand, consistent with the typical mitochondrial gene architecture of teleost fishes (Jing et al. 2024) (Table S3). Overlaps were identified between trnI - trnQ, ATPase 8 - ATPase 6, NAD4L - NAD4, NAD5 - NAD6, and trnT - trnP, with a total overlap length of 21 bp, accounting for approximately 0.13% of the genome. In addition, 13 intergenic spacers with a cumulative length of 60 bp were observed, representing 0.36% of the genome. The mitogenome displayed the highest base composition of adenine (A, 28.5%), followed by cytosine (C, 28.2%), thymine (T, 25.6%), and guanine (G, 17.7%) (Table S4). The 13 PCGs spanned 11,553 bp and encoded 3,851 amino acids. Leucine, isoleucine, threonine, and alanine were the most abundant amino acids (Figure S1). Relative Synonymous Codon Usage (RSCU) analysis (Khandia et al. 2024) revealed a strong preference for codons containing adenine and uracil bases. Several codons, particularly AUU and AAA, had RSCU values > 1, reflecting codon bias within the mitochondrial genome (Table S5).
All PCGs initiated with the start codon “ATG”, except for COX1, which began with “GTG”. The stop codon “TAA” was detected in NAD1, COX1, ATPase 6, NAD4L, and NAD5, whereas “TAG” was used by NAD2, ATPase 8, NAD3, and NAD6. In contrast, COX2, COX3, NAD4, and CYTB terminated with the incomplete stop codon “T–”.
Phylogenetic analysis
Maximum likelihood (ML) and Bayesian inference (BI) trees were constructed using the concatenated sequences of 13 PCGs to determine the evolutionary placement of A. striatus. Both analyses produced congruent topologies. The results indicated that A. striatus and R. shitaiensis shared a closer evolutionary relationship than with other Acheilognathus species. The phylogenetic tree also showed that all Rhodeus species clustered within a single branch, and Rhodeus was more closely associated with Sinorhodeus than with Acheilognathus. Although few genomic studies have been performed on A. striatus, the data obtained here provided important baseline information for future evolutionary research (Figure 3 and 4).
Phylogenetic trees based on ML (upper) and BI (lower) analyses. The accession numbers and citations are as follows: A. barbatulus (NC072602), A. barbatus (NC026872, Tao et al. 2016), A. chankaensis (NC023101, unpublished), A. gracilis (NC028416, unpublished), A. imberbis (NC067619, unpublished), A. majusculus (NC028736, unpublished), A. meridianus (NC031152, unpublished), A. omeiensis (NC037404, unpublished), A. rhombeus (NC028433, unpublished), A. tonkinensis (NC042407, Zhang et al. 2018), B. yodoensis (NC086459, unpublished), G. homalopteroidea (NC085316, unpublished), M. hsinglungshanensis (NC086465, unpublished), P. chii (NC066655, unpublished), R. amarus (NC031538, unpublished), R. cyanorostris (NC060735, Li et al. 2022), R. fangi (NC027437, unpublished), R. notatus (NC029718, unpublished), R. sericeus (NC025326, unpublished), R. shitaiensis (NC022690, Li et al. 2015), S. microlepis (NC042717, Yu et al. 2019), T. lanceolata (NC024566, Xu et al. 2016), T. latimarginata (NC039820, unpublished), T. limbata (NC025515, Luo et al. 2016), T. tanago (NC027665, Miya et al. 2015), T. tinca (NC008648, Saitoh et al. 2006).
Phylogenetic tree based on 13 mitochondrial PCGs(MrBayes Tree).
Discussion and conclusion
The complete mitogenome of A. striatus was successfully obtained and annotated, with a total length of 16,692 bp. Differences in mitogenome length among related Acheilognathinae species may result from variations in tandemly repeated sequences (Wang et al. 2020). The gene arrangement conforms to the typical pattern observed in Acheilognathinae (Zhu et al. 2021).
Bias and skew of base composition in mitogenome have been proposed to arise due to nucleotide mutational bias during mitogenome replication (Lakshmanan et al. 2015). The mitochondrial genome of A. striatus exhibited AT preference and anti-G bias, which consistent with most teleost fishes (Perna and Kocher 1995). Base composition bias was evident across several regions, with both tRNAs and PCGs showing higher AT than GC content, confirming AT preference observed in other fishes (Sun and Xu 2018). The two rRNAs displayed larger absolute AT skew and smaller absolute GC skew values (Ikemura et al. 2023), indicating strong adenine enrichment. Prior studies suggested that AT skew plays a key role in mitochondrial replication mechanisms (Sahyoun et al. 2014). AT-rich regions are also more flexible, influencing DNA bending and winding, which can affect protein binding. Analysis of base composition across the genome further revealed pronounced anti-G bias in PCGs, followed by rRNA genes, whereas tRNAs exhibited mild G bias.
The phylogenetic tree confirmed that A. striatus is closely related to R. shitaiensis, R. fangi, R. notatus, and R. cyanorostris. The reliability of this result was validated through comparison with previous findings (Li et al. 2022). Morphologically, Yang et al. (2010) classified A. striatus within the genus Acheilognathus, whereas molecular evidence suggests possible affinity with the genus Rhodeus. The lack of a clear demarcation between Rhodeus and Acheilognathus within Acheilognathinae necessitates further molecular and morphological validation.
This study presents the mitogenome sequence and structural characteristics of A. striatus, including its gene composition, codon usage bias, and base composition. These findings provide foundational mitochondrial genomic data for Acheilognathus, offering molecular insights to support species identification (Cermakova et al. 2023) and future evolutionary studies within Acheilognathinae.
Supplementary Material
Figure S1Relative Synonymous Codon Usage (RSCU) of Acheilognathus striatus.jpg
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Cermakova E et al. 2023. Identification of fish species and targeted genetic modifications based on DNA analysis: state of the art. Foods. 12(1):228. 10.3390/foods 1201022836613444 PMC 9818732 · doi ↗ · pubmed ↗
- 2Gautam A. 2022. Phenol-Chloroform DNA Isolation Method. Springer International Publishing. p. 33–39.
- 3Ikemura T, Iwasaki Y, Wada Y, Wada K. 2023. Oligonucleotide skews enable comprehensive and insightful characterization of GC- and TA-skew properties observed throughout the human genome with support of unsupervised AI with reference to gene- and Alu-polarity skews. Gene Rep. 33:101852. 10.1016/j.genrep.2023.101852 · doi ↗
- 4Jin J-J et al. 2020. Get Organelle: a fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 21(1):241. 10.1186/s 13059-020-02154-532912315 PMC 7488116 · doi ↗ · pubmed ↗
- 5Jing Y et al. 2024. Influence of life-history traits on mitochondrial DNA substitution rates exceeds that of metabolic rates in teleost fishes. Curr Zool. 71(3):zoae 045–294. 10.1093/cz/zoae 045PMC 1222742540620590 · doi ↗ · pubmed ↗
- 6Kalyaanamoorthy S, Minh BQ, Wong Thomas KF, von Haeseler A, Jermiin LS. 2017. Model Finder: fast model selection for accurate phylogenetic estimates. Nat Methods. 14(6):587–589. 10.1038/nmeth.428528481363 PMC 5453245 · doi ↗ · pubmed ↗
- 7Khandia R, Gurjar P, Kamal MA, Greig NH. 2024. Relative synonymous codon usage and codon pair analysis of depression associated genes. Sci Rep. 14(1):3502. 10.1038/s 41598-024-51909-838346990 PMC 10861588 · doi ↗ · pubmed ↗
- 8Lakshmanan LN, Gruber J, Halliwell B, Gunawan R. 2015. Are mutagenic non D-loop direct repeat motifs in mitochondrial DNA under a negative selection pressure? Nucleic Acids Res. 43(8):4098–4108. 10.1093/nar/gkv 29925855815 PMC 4417187 · doi ↗ · pubmed ↗