Global population structure of Shiga toxin-producing Escherichia coli O103:H2 and the variation in their major virulence factor-encoding genetic elements
Itsuki Taniguchi, Yo Morimoto, Yoko Kimura, Junji Seto, Yuko Kawai, Tomoko Kitahashi, Junko Aoki, Katsuya Terai, Toshihiko Furuta, Yuki Wakabayashi, Sumiko Tanabe, Mitsuhiro Hamasaki, Yuri Abe, Mari Sasaki, Hiroshi Narimatsu, Eiji Yokoyama, Sunao Iyoda, Tetsuya Hayashi

TL;DR
This study explores the global genetic diversity and virulence factors of a harmful type of E. coli called STEC O103:H2, revealing its population structure and evolutionary changes in key disease-causing genes.
Contribution
The study provides the first comprehensive whole-genome analysis of STEC O103:H2, revealing its population structure and genetic variation in virulence elements.
Findings
O103:H2 STEC strains are divided into three distinct lineages based on sequence type.
The major lineage is further split into five clades with C1 as the ancestral group.
Significant genetic variation was found in virulence-related elements like the Stx1a phage and virulence plasmid.
Abstract
Shiga toxin (Stx)-producing Escherichia coli (STEC) is a major cause of serious gastrointestinal illness, including diarrhoea, haemorrhagic colitis and life-threatening haemolytic-uraemic syndrome. Although O157:H7 STEC strains are the most prevalent, the incidence of STEC infections caused by several other serotypes has recently increased. O103:H2 STEC is one of these major non-O157 STEC strains, but systematic whole-genome sequence (WGS) analyses have not yet been conducted. To gain a global phylogenetic overview of O103:H2 STEC based on WGSs, we analysed 2,701 WGSs of O103:H2 strains, including 193 sequenced in this study. Sequence type (ST)-based classification divided the O103:H2 strains into three distinct E. coli lineages. As the virulence marker genes of typical STECs (stx, eae and ehxA) were found only in the major O103:H2 lineage (n=2,658) comprising ST17 and its single- and…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
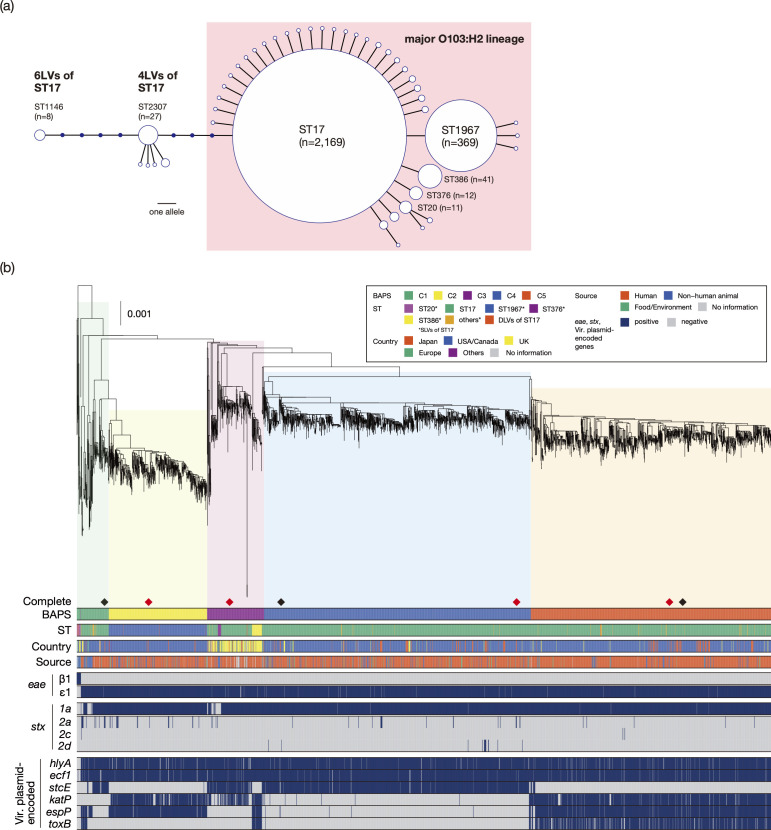 Fig. 1
Fig. 1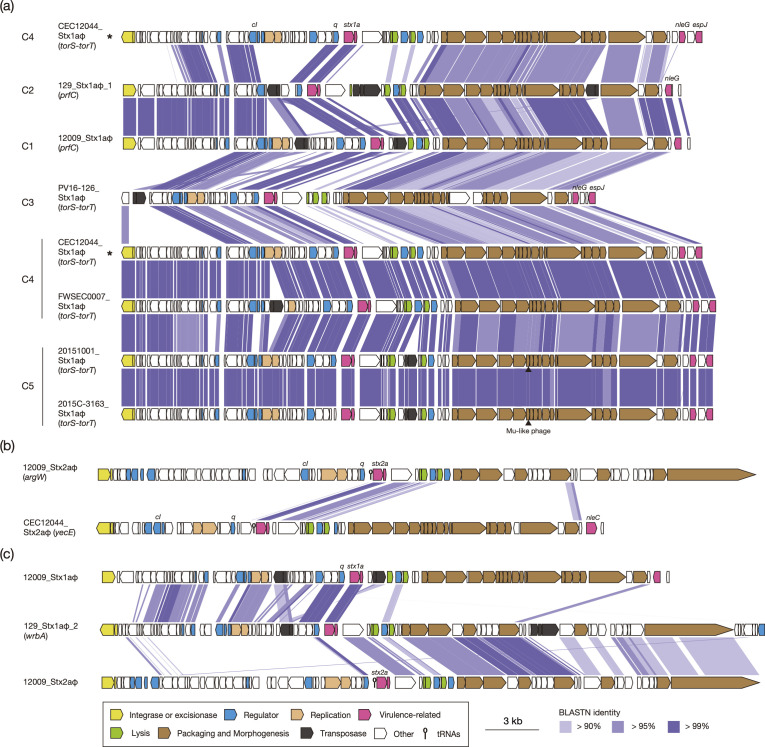 Fig. 2
Fig. 2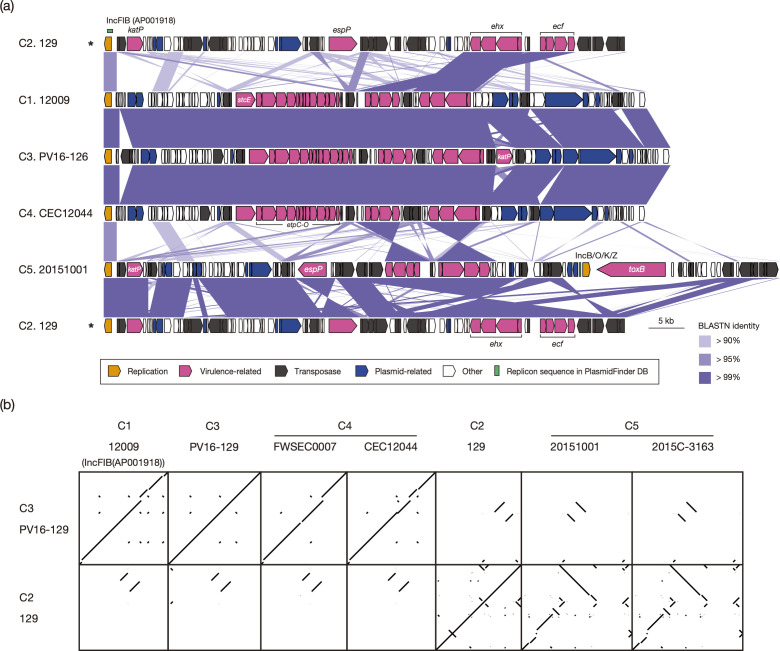 Fig. 3
Fig. 3| Country | Source | Total | |||
|---|---|---|---|---|---|
| Human | Animals | Foods/environments | No information | ||
| Japan | 197 | 3 | 0 | 0 | 200 |
| USA/Canada | 1,814 | 296 | 100 | 10 | 2,220 |
| UK | 178 | 10 | 1 | 13 | 202 |
| Other European countries* | 20 | 26 | 3 | 1 | 50 |
| Other countries† | 6 | 3 | 0 | 0 | 9 |
| No information | 1 | 0 | 0 | 19 | 20 |
| Total | 2,216 | 338 | 104 | 43 | 2,701 |
|
| |||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ST17 and locus variants of ST17 | No. of STs | No. of strains | 1a | 1a×2* | 1c | 1 a/2 a | 1 a/2 c | 1 a/2d | 2a | 2c | 2d | Negative | Epsilon1 | Beta1 | Negative | Positive | Negative |
| ST17 | 1 | 2,169 | 2,061 | 0 | 0 | 32 | 2 | 14 | 13 | 1 | 1 | 45 | 2,137 | 1 | 31 | 2,066 | 103 |
| SLVs | 32 | 482 | 435 | 1 | 0 | 21 | 0 | 0 | 1 | 0 | 0 | 24 | 470 | 11 | 1 | 442 | 40 |
| DLVs | 6 | 7 | 4 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 3 | 4 | 3 | 0 | 3 | 4 |
| 4LV | 5 | 35 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 34 | 0 | 0 | 35 | 0 | 35 |
| 6LV (ST1146) | 1 | 8 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 8 | 0 | 0 | 8 | 0 | 8 |
| Total | 45 | 2,701 | 2,500 | 1 | 1 | 53 | 2 | 14 | 14 | 1 | 1 | 114 | 2,611 | 15 | 75 | 2511 | 190 |
| BAPS | C1 | C2 | C3 | C4 | C4 | C5 | C5 |
|---|---|---|---|---|---|---|---|
| ST | ST17 | ST1967 | ST17 | ST17 | ST17 | ST17 | ST17 |
| Strain | 12009† | 129* | PV16-126* | FWSEC0007† | 20151001* | 2015 C-3163* | |
| Accession no. | AP010958-9 | AP038976-9 | AP038987-8 | CP031908-9 | AP038983-6 | AP038980-2 | CP027219-20 |
| Reference | [ | This study | This study | [ | This study | This study | [ |
| Chromosome (kb) | 5,449 | 5,580 | 5,259 | 5,398 | 5,458 | 5,555 | 5,500 |
| CDSs‡ | 5,254 | 5,443 | 5,003 | 5,148 | 5,250 | 5,365 | 5,309 |
| rRNAs operons | 22 | 22 | 22 | 22 | 22 | 22 | 22 |
| tRNAs | 100 | 100 | 96 | 92 | 95 | 97 | 97 |
| 1a/2a | 1a×2 | 1a | 1a | 1a/2a | 1a | 1a | |
| Plasmid (kb) | 76§ | 73§/73/8 | 79§ | 73§ | 100/75§/2 | 94§/61 | 94§ |
| CDSs | 80 | 82/89/9 | 87 | 74 | 118/83/2 | 97/77 | 100 |
| tRNAs | 0 | 0/0/0 | 0 | 0 | 4/0/0 | 0/0 | 0 |
| Total genome size (kb) | 5,525 | 5,734 | 5,337 | 5,471 | 5,636 | 5,711 | 5,594 |
- —http://dx.doi.org/10.13039/501100001691 Japan Society for the Promotion of Science
- —http://dx.doi.org/10.13039/100009619 Japan Agency for Medical Research and Development
- —http://dx.doi.org/10.13039/100009619 Japan Agency for Medical Research and Development
- —http://dx.doi.org/10.13039/100009619 Japan Agency for Medical Research and Development
- —http://dx.doi.org/10.13039/100009619 Japan Agency for Medical Research and Development
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsEscherichia coli research studies · Vibrio bacteria research studies · Viral gastroenteritis research and epidemiology
Data Summary
The finished genomes of four strains and the Illumina read sequences of 203 O103:H2 strains obtained in this study have been deposited in DDBJ/EMBL/GenBank under BioProject accession numbers starting from PRJDB19807 (https://www.ncbi.nlm.nih.gov/bioproject, see Table S1, available in the online Supplementary Material, for the accession numbers of each strain).
Introduction
Shiga toxin (Stx)-producing Escherichia coli (STEC) are foodborne pathogens that cause a range of diseases, ranging from mild enteritis to severe haemorrhagic colitis and sometimes life-threatening complications, such as haemolytic uraemic syndrome [12]. The major virulence factors of STEC are Stxs encoded by prophages (Stx phages). Stxs are classified into Stx1 and Stx2, and both include multiple variants (Stx1a, Stx1c-Stx1e; Stx2a-Stx2o) [35]. Typical STECs responsible for human infections share the locus of enterocyte effacement (LEE), which encodes a type III secretion system (T3SS), with enteropathogenic E. coli (EPEC) [6], and more than 30 genes encoding effectors have been carried into STEC and EPEC by multiple prophages [79]. Therefore, EPEC strains are generally regarded as progenitors of typical STEC strains. In addition, most STEC strains carry a large virulence plasmid, which harbours an incFIB replicon [10] and encodes enterohaemolysin and other potential virulence factors.
Among various STEC serotypes, O157:H7 is most prevalent worldwide, but strains of other serotypes also cause outbreaks and sporadic cases of infection. O103:H2 is one of the major non-O157 STEC serotypes that causes severe diseases [1113] and is frequently isolated from humans and cattle [1416]. The majority of O103:H2 strains harbour stx1a alone, and isolates carrying stx2a are rarely found [141718]. While the eae and ehxA genes (the marker genes for LEE and virulence plasmids, respectively) are well conserved in O103:H2 strains, virulence plasmid-encoded genes other than ehxA, such as katP (catalase), espP (serine protease) and etp (a type II secretion system gene), are variably distributed [1417].
Whole-genome sequence (WGS)-based phylogenetic analysis of O103:H2 at the global level has not been conducted. Recently, the WGS-based population structure of clonal complex 17 (CC17), which includes O103:H2 strains (more than half of a total of 420 isolates), has been published, but the analysis was limited to strains isolated in England and Wales [19]. In the present study, to analyse the global population structure and evolutionary history of STEC O103:H2, the major serotype within CC17, we collected and analysed 2,701 WGSs of O103:H2 strains, which included 193 WGSs determined in this study, and found that the O103:H2 strains can be classified into three distinct E. coli lineages and that the STEC virulence genes are distributed in the major lineage. We therefore further performed a WGS-based phylogenetic analysis of the O103:H2 strains belonging to the major lineage (n=2,658) and in-depth analyses of the Stx phages, LEE and plasmids in seven closed genomes.
Methods
Bacterial strains
In this study, we sequenced 203 O103:H2 strains isolated in Japan, all of which were human isolates. Of these 203 sequences, 2 were not properly assembled and 8 were low-quality (completeness <99% or contamination >1%, as determined by CheckM [20]); thus, these sequences were excluded. Of the remaining 193 WGSs of Japanese strains, four were closed as described below. To collect O103:H2 WGS data available in public databases, we downloaded O103:H2 read or assembled sequence data from the National Center for Biotechnology Information (NCBI) database and O103:H2 assembled sequences from EnteroBase (n=3013, access date 28 February 2022). After confirming their serotypes using Abricate (https://github.com/tseemann/abricate) with the EcOH database [21] and excluding low-quality sequences with the same threshold as that described above, 2,508 genomes were included in the dataset. Thus, a total of 2,701 O103:H2 strains were analysed (listed in Tables 1 and S1). Their sequence types (STs) were determined by a blastn-based strategy using the mlst program v2.23.0 (https://github.com/tseemann/mlst) with Achtman’s scheme and the PubMLST database (https://pubmlst.org). Genomes whose STs were not precisely defined were reanalysed by a read mapping-based strategy using the SRST2 program v0.2.0 [22] with the default parameters. Relationships between the identified STs were analysed via the MSTree V2 algorithm packaged in the GrapeTree program [23], and a minimum spanning tree (MST) based on the STs was visualized using the same program.
Genome sequencing, assembly and annotation
The purification of genomic DNA, preparation of sequencing libraries, Illumina MiSeq sequencing and sequence assembly were performed as previously described [24]. The average sequencing depth and number of scaffolds were 47×‒378× and 187‒850, respectively. Four strains (129, PV16-126, CEC12044 and 20151001) were additionally sequenced using MinION with R10.4.1(129 and PV16-126) or R9.4.1 (CEC12044 and 20151001) flow cells [Oxford Nanopore Technologies (ONT)] for 72 h (129 and PV16-126) or 96 h (CEC12044 and 20151001). Read data in fastq format were generated using MinKNOW v23.4.8 and Guppy v6.5.7 (129 and PV16-126) or MinKNOW v3.1.13 and qcat v1.1.0 (CEC12044 and 20151001). These reads were trimmed and filtered using the following programme and parameters: trimming using Porechop (v0.2.4) [25] and filtering over 2 kb at a quality score (Q score) > 8 using NanoFilt (v2.8.0) [26], with the option of trimming 100 bp from the start of the read in strains 129 and PV16-126, or filtering over 2 kb at a Q score >10 using NanoFilt (v2.3.0), with the option of trimming 100 bp from the start of the read in strains CEC12044 and 20151001. The trimmed and filtered ONT and Illumina reads of each strain were assembled using the MicroPIPE pipeline [27] for strains 129 and PV16-126 or Unicycler v0.4.7 [28] for strains CEC12044 and 20151001. A tandemly duplicated genome generated for a small plasmid (~8 kb) in strain 129 was manually corrected. The closed genomes were annotated using DFAST [29], followed by manual curation. GenomeMatcher (v3.0.8) [30] was used for genome sequence comparison and dot-plot analysis and to display the results. The presence of plasmid replicons was determined using PlasmidFinder v.2.1 [31] with default parameters.
SNP detection and phylogenetic analysis
Phylogenetic analyses were performed for the O103:H2 strain set comprising strains belonging to ST17 (n=2,169) and its single-locus variants (SLVs) and double-locus variants (DLVs) (482 and 7, respectively) (Tables 2 and S1) using the O103:H2 strain 12009 as a reference with or without the O26:H11 strain 11368 [9]. For these analyses, SNP sites on prophage (PP)/integrative element (IE)/insertion sequence (IS)-free and recombination-free chromosome backbone sequences conserved in all analysed genomes were identified using MUMmer [32] and Gubbins [33] to construct maximum likelihood (ML) trees using RAxML as previously described [34]. For tree construction, strains were deduplicated if the chromosome backbone sequences were identical. Clustering analysis of O103:H2 strains was performed using Fastbaps v1.0.8 with ‘BAPS’ [35], and clades were defined at the first level of hierarchical clustering. ML trees were displayed using iTOL [36].
Analysis of the stx, eae and ehxa genes
The subtypes of stx and eae were determined by blastn as previously described [37], but with slightly different thresholds (>98% identity and >50% coverage for stx and >99% identity and >90% coverage for eae). Plasmid-encoded virulence genes (ehxA, ecf1, stcE, katP, espP and efa1) were also identified by a blastn search (>98% identity and >30% coverage), with the representative sequences of each gene used as references (listed in Table S2). As both stx and plasmid-encoded virulence genes were sometimes fragmented in draft genomes, we used a lower threshold than that applied to the eae gene.
Results and discussion
Strain set
We analysed a total of 2,701 O103:H2 genome-sequenced strains in this study. These strains included 193 Japanese strains sequenced in this study (all human isolates) and 2,508 strains whose genomes were obtained from public databases and were isolated in various geographical regions (Tables 1 and S1). North American (USA/Canada) strains represented 82 % of the strain set, followed by strains from the UK (n=202) and Japan (n=200). Although most strains (n=2,216) were human isolates, isolates from nonhuman animals (bovine, avian, swine and others; n=338) and foods/environments (beef, water, plant and others; n=104) were also included.
A total of 45 STs were identified in the O103:H2 strain set. While the majority of strains (n=2,169) belonged to ST17 and most of the remaining strains (n=489) belonged to the SLVs and DLVs of ST17 (32 and 6 STs, respectively), five STs were 4-locus variants (4LVs) and one was a 6-locus variant (6LV) of ST17 (Table 2). As the 4LV and the 6LV differed at five loci, this analysis revealed the presence of three distinct E. coli lineages with the O103:H2 serotype (Fig. 1a). Notably, none of the 4LV and 6LV strains (n=43) contained any of the marker genes of major STEC lineages (stx, eae and ehxA), except for one strain containing the stx1c gene alone. In the following analyses, we analysed the strains belonging to ST17 and its SLVs and DLVs (referred to as the major O103:H2 lineage; n=2,658). Among the 2,658 strains belonging to the major O103:H2 lineage, 2,169 and 369 strains belonged to ST17 and ST1967, respectively (Table S3). Examination of the distribution of major STEC virulence-related genes (stx1, stx2, eae and ehxA) revealed that while the stx1a, eae and ehxA genes were present in almost all strains, stx2 was detected in a limited number of strains (Table 2).
Relationships among the E. coli O103:H2 strains analysed in this study. (a) An MST displaying the population structure of 2701 O103:H2 strains based on allele sequence combinations of the adk, fumC, gyrB, icd, mdh, purA and recA genes. The sizes of the open circles representing each ST are scaled to the number of isolates belonging to each ST. Hypothetical STs not found in the strain set are represented by filled circles. The O103:H2 major lineage, to which ST17 and its SLVs and DLVs belong, is highlighted. (b) Phylogenetic relationships of 2,497 strains belonging to the major O103:H2 lineage. An ML tree constructed based on 29,938 SNPs identified on the PP/IE/IS-free and recombination-free chromosomal backbone (2,045,250 bp) is shown along with strain information on Fastbaps clades (BAPS), STs, isolation countries and sources, the presence/absence of eae (subtypes β1 and ε1), stx (stx1a, stx2a, stx2c and stx2d) and the STEC virulence plasmid (Vir. Plasmid)-encoded virulence-related genes. Strains belonging to ST20 and its SLVs are used as outliers according to the results shown in Fig. S1. The five BAPS clades are highlighted in different colours in the tree. The positions of the seven closed-genome strains are indicated by diamonds. Among the seven strains, four strains sequenced in this study are indicated by red diamonds. Bar, the mean number of nucleotide substitutions per site.
Phylogenetic and evolutionary overview of the major O103:H2 lineage
The 2658 strains belonging to the major O103:H2 lineage were isolated in 19 countries. We first constructed a WGS-based ML tree of these strains with the O26:H11 strain 11368 as an outlier and found that ST20 and its SLVs (ST1790 and ST9133; both were DLVs of ST17; Fig. 1a, Table S3) were first separated from the major O103:H2 lineage (Fig. S1). Therefore, we used the strains belonging to ST20 and its SLVs as outliers to construct the ML tree of the major O103:H2 lineage (Fig. 1b). Among the 2,658 genomes analysed, 249 were identical to one or more genomes in terms of their backbone chromosome sequence (88 groups; Table S1). To reduce strain redundancy, one representative strain was selected from each group and included in the following analyses. WGS-based phylogenetic analysis of this strain set (n=2497) revealed five distinct clades (C1–C5), as defined by Fastbaps-based clustering (Fig. 1b). Although the strain set included relatively small numbers of strains from nonhuman sources, these strains were distributed between human strains in each clade. The results of this phylogenetic analysis suggest that C1 (comprising mainly ST17 strains but including ST20 and other minor ST strains) was ancestral in the major O103:H2 lineage and that C2 (ST1967) and C3 (ST17, ST376 and ST386) emerged from C1, after which C4 (ST17) and C5 (ST17) emerged from C3. Notably, ST20 and its SLVs harboured neither stx nor ehxA. Although they contained the eae gene, its subtype (beta1) differed from that (epsilon1) of other strains in C1 and the other clades, indicating that the strains belonging to ST20 and its SLVs included in the strain set analysed in this study are EPEC and that an eae subtype change occurred upon the separation of the ST20 lineage from the other strains in the major O103:H2 lineage. Hereafter, strains other than the strains belonging to ST20 and its SLVs (n=2483) are referred to as STEC O103:H2.
Among the five clades, strains from the USA/Canada, showing the highest proportion in the current dataset (82%), presented the highest proportion in four of the clades (C1, C2, C4 and C5; 74–96%) (Fig. 1b; see Table S4 for details). However, most C3 strains (74%) were isolated in European countries (mainly from the UK but also other European countries; Table S4); thus, C3 is the clade circulating mainly in Europe. Analysis of the distribution of the major STEC virulence-related genes in the STEC O103:H2 strains revealed that while stx1a, eae (subtype epsilon1) and ehxA were well conserved, the stx2a, stx2c and stx2d genes were present in small numbers of strains (n=67, 3 and 15, respectively) (Fig. 1b, Table 2). Although the distributions of stx2c and stx2d were very limited to C1 and C3 (stx2c) and C4 (stx2d), stx2a was distributed in all clades but was distributed sporadically.
General genomic features of STEC O103:H2 inferred from closed genomes
While three closed genome sequences are publicly available [93839], they belong to three of the five clades (C1, C4 and C5) (Table 3). To gain a wider genomic view of STEC O103:H2, particularly on Stx phages, the LEE and the STEC virulence plasmids, we determined the complete genome sequences of two strains belonging to C2 and C3, respectively, and two additional strains belonging to C4 and C5, respectively. As summarized in Table 3, the chromosomes of the seven strains were 5,259–5,580 kb in size. The chromosome backbone was well conserved between the strains, although many indels, such as the loss of Stx2a phage, were observed between the strains (Fig. S2). A small inversion occurred in the strains belonging to C3, C4 and C5 (Fig. S2). In particular, the chromosomes of the strain pairs belonging to the same clade were highly conserved (C4 and C5), even though the two C4 strains were distantly related in this clade. All strains carried an STEC virulence plasmid, and three strains contained one or two additional plasmids (see the next subsection for the details of these plasmids).
Variation in Stx phages, the LEE and plasmids in the closed STEC O103:H2 genomes
Stx phages
Of the seven strains with fully sequenced genomes (closed-genome strains), four contained an Stx1a phage alone, the strain belonging to C1 (strain 12009) and one of the two C4 strains (strain CEC12044) contained Stx1a and Stx2a phages and the strain belonging to C2 (strain 129) carried two Stx1a phages (Table 3). Among the eight Stx1a phages identified in the seven genomes, seven were long-tailed phages with a set of late genes similar to that of phage lambda (Fig. 2a), and the remaining phage, the second Stx1a phage in strain 129, was a short-tailed phage encoding a set of late genes similar to that of the Stx2a phages of O157:H7 STEC strains [4041] (Fig. 2c).
Comparison of the Stx phages identified in the seven closed-genome strains. Genomic organizations of seven long-tailed Stx1a phages (a), two Stx2a phages (b) and the short-tailed Stx1a phage of strain 129 and the long-tailed Stx1a phage and the short-tailed Stx2a phage of strain 12009 (c) are drawn at scale. The Stx1a phage of strain CEC12044, which is illustrated twice, is indicated by asterisks. The levels of nucleotide sequence identities between homologous coding sequences (CDSs) are indicated by heatmaps.
The seven long-tailed Stx1a phages were 45.3–56.4 kb in size, but a transposable Mu-like phage (39.3 kb) [42] was integrated into the phages of both C5 strains (20151001 and 2015 C-3163) (Fig. 2a). Two loci (prfC and the torS-torT intergenic region) were identified as the integration sites of these Stx1a phages: the Stx1a phages of the C1 and C2 strains were found at prfC, and those of the C3, C4 and C5 strains were at torS-torT. While these seven Stx1a phage genomes presented overall sequence similarities, the sequences of the region including the int gene in the Stx1a phages of the C1 and C2 strains were different from those of the C4 and C5 strains. Although an IS-associated deletion occurred in this region in the C3 strain, a fragment of the int gene homologous to those of the C4 and C5 strains remained, suggesting that the Stx1a phages of the C3 strain originally contained the int-containing region similar to those of the C4 and C5 strains. Considering the phylogenetic relationship of the host strains, these findings suggest that genomic recombination inducing the integration site switch from prfC to torS-torT occurred in the Stx1a phage during the evolution of STEC O103:H2. Similar but more dynamic changes in Stx1a phages were observed in the O26:H11 ST21 lineage, including the turnover of Stx1a phage (replacement by apparently different Stx1a phages at the same or different chromosome loci) [43].
The sequences of the two Stx2a phages identified in the C1 and C4 strains were highly divergent: one was a long-tailed phage, and the other was a short-tailed phage (Fig. 2b). Interestingly, the second Stx1a phage of the C2 strain (strain 129) exhibited a chimeric structure with early and late regions similar to those of the long-tailed Stx1a phage and the short-tailed Stx2a phage of the C1 strain (strain 12009), respectively (Fig. 2c).
Locus of enterocyte effacement
LEE was present at pheV in all the closed-genome strains (Fig. S2). The core region encoding a set of T3SS genes was well conserved between the strains, although the integrase gene was degraded by the insertion of IS629 in four strains (PV16-126, FWSEC0007, 20151001 and 2015 C-3163) (Fig. S3). These findings indicate that LEE is maintained at the pheV locus in the STEC O103:H2 lineage. However, various rearrangements, such as deletions and inversions, have occurred in the accessory region where several virulence-related genes, such as the nleE, nleB and espL genes for T3SS effectors, ag43 (also known as flu) for the autotransporter antigen 43 [44] and efa1 for adhesin Efa (E. coli factor for adherence; almost identical to lymphostatin encoded by lifA) [45], are encoded. Many of these rearrangements are also likely induced by IS-related mechanisms.
Plasmids
Virulence plasmids were highly conserved in sequence and gene organization between the C1, C3 and C4 strains (12009, PV16-126, FWSEC0007 and CEC12044), except for the insertion of the katP gene in the C3 strain and the insertion or deletion of a few IS elements (Fig. 3). However, the virulence plasmids of the C2 and C5 strains (129, 20151001 and 2015 C-3163) exhibited marked differences from those of the C1, C3 and C4 strains, and only the regions encoding the replication gene and the ehx and ecf operons were shared. Although the backbone sequences of the plasmids of the C2 and C5 strains were similar, marked structural variations due to IS-related deletions and inversions were observed between them. In addition, the toxB gene encoding an adhesin [46], which was first identified in the virulence plasmid of the O157:H7 strain Sakai (pO157) [47], was encoded by only the plasmids of the C5 strains. The two C5 plasmids contained the IncB/O/K/Z replicon in addition to the IncFIB (AP001918) replicon which was shared by all virulence plasmids (Fig. 3a). These results suggest that while the virulence plasmid acquired by the common ancestor of STEC O103:H2 has been maintained in C1, C3 and C4, it was replaced by other plasmids in C2 and C5. Although the virulence plasmids of the C2 and C5 strains presented some similarity, they were likely acquired independently.
Variations in the STEC virulence plasmids among the seven closed-genome STEC O103:H2 strains. (a) Genomic organizations of the STEC virulence plasmids identified in the seven closed-genome strains. The levels of nucleotide sequence identity between homologous coding sequences (CDSs) are indicated by a heatmap. ‘Plasmid-related’ CDSs include genes for conjugation, mobilization, partitioning, maintenance and SOS inhibition. The plasmid of strain 129, which is illustrated twice, is indicated by asterisks. (b) Dot plot showing the sequence similarities (>99% sequence identity) between the virulence plasmids of the seven closed-genome strains. The nucleotide sequences of the virulence plasmids of strains PV16-129 (clade C3) and 129 (clade C2) were compared with those of other strains.
The present genome assemblies inferred the presence of one or two additional plasmids in strains 129 (C2), CEC12044 (C4) and 20151001 (C5) (Fig. S3a). Of these, two plasmids (72.8 and 61.4 kb) carried by strains 129 and 20151001 shared a set of genes for conjugation, stabilization and SOS inhibition, but antimicrobial resistance genes, the streptomycin resistance genes (strAB), the sulphonamide resistance gene (sul) and the TEM-1 β-lactamase gene (blaTEM-1B), were found only in the plasmid of strain 129. For the large plasmid (101 kb) carried by strain CEC12044, the functions of most genes are unknown, except for several genes, such as three prophage-related genes, a DNA polymerase theta subunit-encoding gene and two tRNA genes.
Variation in the plasmid-encoded virulence-related gene repertoire among the STEC O103:H2 strains
As the virulence-related genes of the virulence plasmids of the closed-genome strains showed notable variation, we analysed the distribution of six plasmid-encoded virulence-related genes in the major O103:H2 lineage (Fig. 1b; note that the ST20 strains and their close relatives lacked not only ehxA but also the other five genes). Among the six genes, ehxA and ecf1, which were found in the virulence plasmids of all the closed-genome strains, were well conserved in the entire STEC O103:H2 lineage. High conservation of these genes has also been observed in other STEC lineages, such as STEC O26:H11, O121:H19 and O145:H28, as well as in STEC belonging to clonal complex 119 (CC119; O165:H25 and O172:H25) [24343748], suggesting the biological importance of these two genes (or the ehx and ecf operons) in STEC. The results of the analysis of STEC O103:H2 obtained in this study provide further support for this notion. The remaining four genes (stcE, katP, espP and toxB) presented distribution patterns unique to each clade. In C1, while strains belonging to early separated branches contained espP and toxB or espP alone, close relatives of strain 12009 contained only the stcE gene, as observed for this closed-genome strain. In C2, most strains contained katP and espP, as observed for the closed-genome strain (129). In C3, most strains contained stcE and katP, as observed for the closed-genome strains (PV16-126), but strains belonging to one sublineage (ST386 strains) contained katP, espP and toxB. In C4, while katP and espP were sporadically found in several strains, most strains contained only stcE, as observed for the closed-genome strains (FWSEC0007 and CEC12044). In C5, most strains contained katP, espP and toxB, as observed for the closed-genome strains (201510001 and 2015 C-3168), but several strains that were separated early from the other strains in clade C5 contained stcE and katP. These findings suggest that the repertoires of virulence-related genes on the virulence plasmids in each clade were largely consistent with those identified in the closed-genome strains belonging to each clade, but there were considerable intraclade variations. The abundance of IS elements on the virulence plasmid genomes may be related to the generation of such intraclade variations.
Conclusion
In this study, we analysed the WGS data for 2701 O103:H2 strains isolated from various geographic regions, including the 193 Japanese strains sequenced in this study. These strains belong to three distinct E. coli lineages, and the virulence marker genes for typical STEC (eae, stx and ehxA) are found only in the major lineage (ST17 and its SLVs and DLVs; called the major O103:H2 lineage in this manuscript). We defined five clades (C1-C5) in the major O103:H2 lineage, of which C1 was the ancestral clade, C2 and C3 emerged from C1 and C4 and C5 emerged from C3. As ST20 and its SLVs found in clade C1 did not contain stx and ehxA and the subtype of their eae genes was different from that of the other strains, the globally circulating STEC O103:H2 lineage emerged by acquiring stx and ehxA through the acquisition of phages and plasmids encoding these genes after separating from ST20 and its SLVs. A change in the eae subtype also occurred in this process. While stx2a, stx2c and stx2d were sporadically distributed in limited STEC O103:H2 strains, stx1, eae (epsilon1 subtype) and ehxA were highly conserved in the entire STEC O103:H2 lineage. However, detailed analyses of the closed genomes of seven STEC O103:H2 strains covering the five clades revealed marked variations in the genetic elements encoding these genes, such as rearrangements in the LEE accessory region and a shift in the integration sites of the long-tailed Stx1a phage due to the replacement of the int-containing genomic segments. Marked genomic diversity was also observed for the virulence plasmids, which were generated by the replacement of plasmids and the gain and loss of virulence-related genes, the latter of which was likely related to IS elements that abundantly occur in virulence plasmids. These results provide the current global phylogenetic overview of O103:H2 strains and expand our understanding of the variation in the major virulence determinants within STEC O103:H2, which is relatively understudied among the major STEC lineages.
Supplementary material
10.1099/mgen.0.001625Uncited Supplementary Material 1.
10.1099/mgen.0.001625Uncited Supplementary Material 2.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Ylinen E Salmenlinna S Halkilahti J Jahnukainen T Korhonen L et al Hemolytic uremic syndrome caused by Shiga toxin-producing Escherichia coli in children: incidence, risk factors, and clinical outcome Pediatr Nephrol 2020351749175910.1007/s 00467-020-04560-032323005 PMC 7385025 · doi ↗ · pubmed ↗
- 2Dato L Mancuso MC Daprai L Ria T Rossetti D et al Bloody diarrhea, STEC infection, and HUS in the molecular microbiology era Pediatr Nephrol 202510.1007/s 00467-025-06930-y PMC 1295346040848063 · doi ↗ · pubmed ↗
- 3Scheutz F Teel LD Beutin L Piérard D Buvens G et al Multicenter evaluation of a sequence-based protocol for subtyping Shiga toxins and standardizing Stx nomenclature J Clin Microbiol 2012502951296310.1128/JCM.00860-1222760050 PMC 3421821 · doi ↗ · pubmed ↗
- 4Probert WS Mc Quaid C Schrader K Isolation and identification of an Enterobacter cloacae strain producing a novel subtype of Shiga toxin type 1J Clin Microbiol 2014522346235110.1128/JCM.00338-1424759708 PMC 4097712 · doi ↗ · pubmed ↗
- 5Lindsey RL Prasad A Feldgarden M Gonzalez-Escalona N Kapsak C et al Identification and characterization of ten Escherichia coli strains encoding novel shiga toxin 2 Subtypes, Stx 2n as well as Stx 2j, Stx 2m, and Stx 2o, in the United States Microorganisms 202311256110.3390/microorganisms 1110256137894219 PMC 10608928 · doi ↗ · pubmed ↗
- 6Croxen MA Law RJ Scholz R Keeney KM Wlodarska M et al Recent advances in understanding enteric pathogenic Escherichia coli Clin Microbiol Rev 20132682288010.1128/CMR.00022-1324092857 PMC 3811233 · doi ↗ · pubmed ↗
- 7Deng W Puente JL Gruenheid S Li Y Vallance BA et al Dissecting virulence: systematic and functional analyses of a pathogenicity island Proc Natl Acad Sci USA 20041013597360210.1073/pnas.040032610114988506 PMC 373508 · doi ↗ · pubmed ↗
- 8Tobe T Beatson SA Taniguchi H Abe H Bailey CM et al An extensive repertoire of type III secretion effectors in Escherichia coli O 157 and the role of lambdoid phages in their dissemination Proc Natl Acad Sci USA 2006103149411494610.1073/pnas.060489110316990433 PMC 1595455 · doi ↗ · pubmed ↗