A review of neurophysiological relationships between sleep disorders and depression
Yifan Huang

TL;DR
This review explores how sleep disorders and depression share common neurophysiological mechanisms, offering insights into potential new treatments.
Contribution
The paper introduces astrocytic dysfunction and GABA/glutamate imbalance as novel mechanisms linking sleep disorders and depression.
Findings
Shared mechanisms include monoamine, HPA axis, and inflammatory factor dysregulation.
Astrocytic dysfunction and GABA/glutamate imbalance reinforce pathological feedback loops.
Treating sleep disorders may improve depression outcomes through shared therapeutic targets.
Abstract
This review critically synthesizes current neurophysiological findings on the comorbidity between depression and sleep disorders. Drawing upon a multidisciplinary body of literature, the paper delineates overlapping neurochemical, hormonal, and inflammatory mechanisms. Further, it explores the role of astrocytic dysfunction and glutamate-GABA imbalance in reinforcing pathological feedback loops. By adopting an integrative framework, this review underscores the bidirectional and systemic nature of sleep disorder-depression comorbidity, offering insights into shared pathophysiological substrates and potential therapeutic targets for future research. •The neurophysiological mechanisms of sleep-depression comorbidity are systematically reviewed.•A framework integrating monoamine, HPA axis, and inflammatory factor dysregulation is proposed.•Astrocytic dysfunction and GABA/glutamate…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
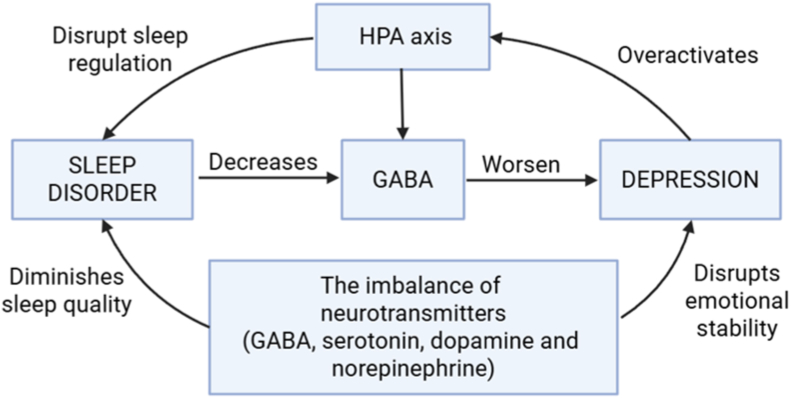 Figure 1
Figure 1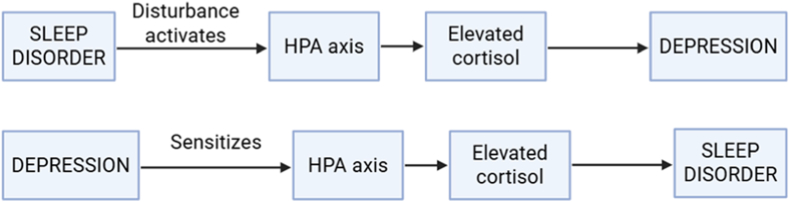 Figure 2
Figure 2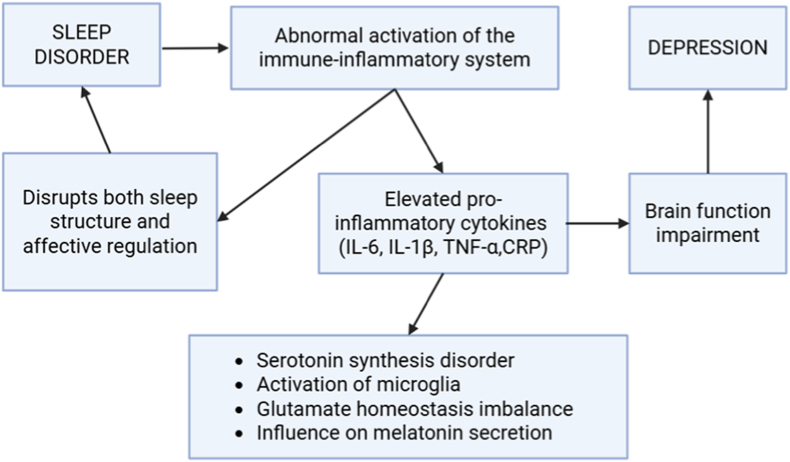 Figure 3
Figure 3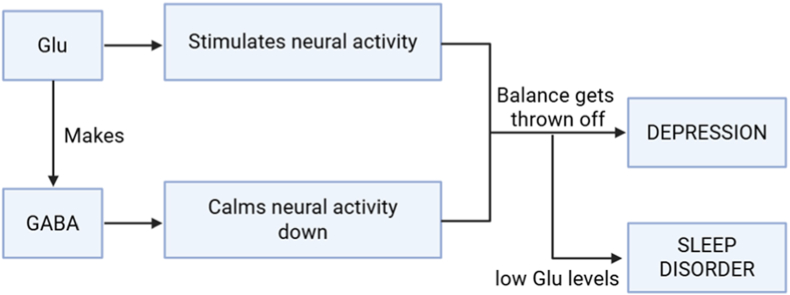 Figure 4
Figure 4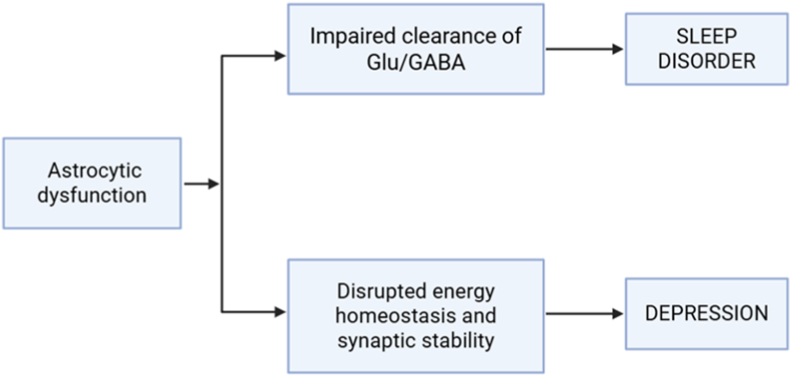 Figure 5
Figure 5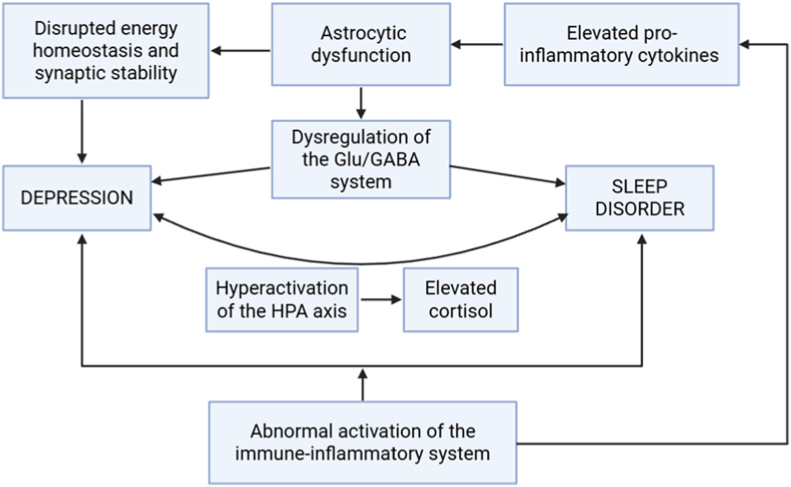 Figure 6
Figure 6Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsSleep and Wakefulness Research · Sleep and related disorders · Tryptophan and brain disorders
Introduction
1
Sleep is a fundamental physiological process essential for physical health and psychological well-being. It is regulated by complex neural systems that coordinate transitions between wakefulness and sleep stages through intricate circuitry and neurotransmitter dynamics (Saper and Fuller, 2017). Sleep disorders encompass a range of conditions that disrupt normal sleep architecture, including obstructive sleep apnea, insomnia, reduced sleep duration, restless legs syndrome, and narcolepsy (Chen et al., 2014). These disorders may arise as primary conditions due to physiological dysfunction or maladaptive behaviors (Harvey, 2001) and are associated with adverse outcomes such as impaired glucose regulation, cardiovascular dysfunction, emotional dysregulation, and cognitive deficits (Amagai et al., 2010; Beihl et al., 2009; Chen et al., 2014; Itani et al., 2011).
Depression and sleep disorders exhibit a robust bidirectional relationship, and this association extends beyond insomnia to include other sleep disturbances such as hypersomnia, obstructive sleep apnea (OSA), and restless legs syndrome, all of which are significant prospective risk factors for depression (Germain & Kupfer, 2008; Kendzerska et al., 2014; Winkelman et al., 2008; Zhang et al., 2022). Among sleep disorders, insomnia is the most prevalent and is closely linked to depression. It serves not only as a symptom but also as a significant risk factor and early predictor of depressive episodes (Fernandez-Mendoza and Vgontzas, 2013; Otsuka et al., 2017). As evidenced by meta-analyses indicating that individuals with insomnia face a significantly increased risk of developing major depressive episodes (OR = 2.30) (Zhang et al., 2022). Besides, longitudinal studies indicate that insomnia increases the likelihood of developing depression (Baglioni et al., 2011), and its severity correlates with depressive symptoms (Riemann et al., 2001). Notably, treating insomnia can reduce depressive symptoms, suggesting a potential causal relationship (Manber et al., 2008).
Depression is a prevalent psychiatric disorder characterized by emotional, cognitive, and somatic symptoms, including persistent low mood, anhedonia, negative thoughts, cognitive impairment, appetite and sleep changes, fatigue, and physical pain (Harm et al., 2013; Malhi and Mann, 2018). Its lifetime prevalence is estimated at 11–17 % globally (Steinert et al., 2014), with peak vulnerability during adolescence, early adulthood, and late adulthood (Malhi et al., 2018). Neurobiologically, depression involves monoaminergic dysregulation, hypothalamic-pituitary-adrenal axis (HPA axis) hyperactivity, neuroinflammation, reduced neurotrophic signaling (e.g., serum brain-derived neurotrophic factor), and altered cortical–subcortical circuit function (Li et al., 2021; Plante, 2021). These mechanisms are also implicated in sleep regulation, suggesting shared neurophysiological foundations.
The pathophysiology underlying this comorbidity involves dysregulation of neuroplasticity, emotional processing, and the hypothalamic-pituitary-adrenal axis, with circadian rhythm disruptions playing a critical role (Drakatos et al., 2023; Liguori et al., 2021). Furthermore, specific patient populations, including those with Post COVID-19 syndrome and autoimmune diseases like primary Sjogren's syndrome, demonstrate a high prevalence of concurrent depression, anxiety, and sleep disorders, highlighting the shared mechanisms and substantial burden of these co-occurring conditions (Han et al., 2025; Seighali et al., 2024).
Recent research has identified potential biomarkers, such as decreased serum brain-derived neurotrophic factor (BDNF) levels, which are negatively correlated with poor sleep quality and demonstrate predictive value for sleep disorders in clinical populations (Han et al., 2025). The recognition of this strong interconnection necessitates integrated clinical approaches, including routine depression screening in sleep centers and sleep assessment in psychiatric settings, to facilitate early detection and tailored interventions (Daccò et al., 2025).
Despite extensive correlational data, few studies have systematically examined the neural substrates common to both conditions. This review addresses this gap by integrating neurophysiological research from EEG, neuroimaging, and neurochemical studies, with a focus on mechanistic insights underlying comorbidity.
Methods
2
This review was conducted as a narrative, mechanism-oriented synthesis and followed the SANRA (Scale for the Assessment of Narrative Review Articles) guidelines to ensure methodological transparency, coherence of scientific reasoning, and critical integration of heterogeneous evidence. Literature was identified through structured searches of PubMed, Embase, Web of Science, and the Australian National University Library databases, covering publications from 2000 to 2025. The literature search was restricted to studies published between 2000 and 2025 to ensure methodological consistency, clinical relevance, and alignment with contemporary research standards. Since 2000, substantial advances have occurred in diagnostic criteria for depression, sleep assessment techniques (e.g., standardized questionnaires and objective measures), and neurobiological and epidemiological methodologies. Earlier studies often relied on outdated diagnostic frameworks and less rigorous designs, which may limit their comparability with current evidence. Focusing on this time frame allows the review to capture modern theoretical models, robust longitudinal data, and clinically applicable findings, while older studies are considered primarily for historical context rather than as core evidence.
Search terms were used individually and in combination and included “sleep disorders,” “depression,” “comorbidity.” After reviewing additional literature, more keywords were employed for precise screening, including “circadian rhythm,” “GABA,” “glutamate,” “monoamines,” “HPA axis,” “inflammation,” and “astrocytes.” Studies were selected based on thematic relevance to neurophysiological mechanisms linking sleep disturbances and depression, rather than on uniform outcome measures.
See Appendix A Table 2 for an overview of selected studies. Inclusion criteria were: (1) studies published in English; (2) review articles focusing on either sleep disorders, depression, or both; (3) diagnosed depression using DSM-IV or DSM-5 criteria; (4) sleep disorders including insomnia, sleep apnea, circadian disorders, or restless legs syndrome; and (5) inclusion of neurophysiological measures such as EEG, fMRI, hormonal assays, or neurochemical markers.
The inclusion criteria for studies in Appendix B Table 3 were: (1) studies published in English; (2) human sample size ≥10; (3) original researches focusing on either sleep disorders, depression, or both; (4) inclusion of neurophysiological indicators such as circadian rhythms, neurotransmitters (e.g., GABA, serotonin, dopamine), neuroimaging, inflammatory markers, or brain network functions; and (5) exploration of the biological or neurophysiological mechanisms underlying the relationship between sleep and depression.
A standardized quality and risk-of-bias assessment was conducted for all included studies, and summary tables were generated to present the evaluations together with the principal potential sources of bias. Studies were classified by design type and assessed using established, design-specific instruments: animal experiments were evaluated with the SYRCLE risk-of-bias tool; observational studies (cohort, case–control, cross-sectional) with the Newcastle–Ottawa Scale (NOS); randomized controlled trials with the Cochrane Risk of Bias 2 (RoB 2) tool; and systematic reviews and meta-analyses with AMSTAR 2. Appendix C Table 4 provides a template summary table containing, for each reference, the study design, an overall quality or risk rating (or score where applicable), a domain-level evaluation (e.g., selection bias, information bias, confounding), and a concise description of the primary potential sources of bias (such as convenience sampling, inadequate blinding, small sample size, or incomplete adjustment for confounding variables). These standardized assessments were employed to support the interpretation of the findings and to inform the sensitivity analyses.
This review was conducted considering principles from the SANRA guideline. All studies were manually screened to ensure methodological validity and thematic relevance. Articles lacking clear neurophysiological focus, human applicability, or standardized diagnostic criteria were excluded. Priority was given to studies that integrated clinical findings with neurobiological mechanisms, thereby strengthening the analytical foundation for examining the bidirectional relationship between sleep disorders and depression.
Clarifying terminology
3
Sleep disorders vs. insomnia
3.1
While this review focuses on insomnia as a representative case, it is important to distinguish between the broader category of sleep disorders and specific diagnoses. Insomnia is characterized by difficulties in sleep initiation, maintenance, or early-morning awakenings, and is both a symptom and independent disorder. In contrast, other sleep disorders such as obstructive sleep apnea (linked to upper airway collapse and nocturnal hypoxia), restless legs syndrome (marked by limb discomfort and motor restlessness), and circadian rhythm disorders (involving misaligned sleep-wake cycles) manifest distinct pathophysiological and neurophysiological features. Each of these disorders may co-occur with depression and contribute to unique neurochemical imbalances.
SANRA vs. PRISMA
3.2
The SANRA framework was selected in preference to PRISMA because the primary objective of this review was to integrate and interpret diverse neurophysiological evidence rather than to conduct a systematic review or meta-analysis. PRISMA is optimized for quantitatively synthesizing homogeneous studies with comparable designs, populations, and outcome measures. In contrast, the present review encompasses heterogeneous methodologies, including epidemiological studies, neuroimaging, electrophysiology, molecular biomarkers, and preclinical animal research, which are not amenable to pooled statistical analysis. Consequently, formal meta-analytic procedures and fully reproducible search strategies were neither methodologically appropriate nor conceptually aligned with the aims of the study. SANRA provides a more suitable framework by emphasizing clarity of research rationale, transparency of literature selection, critical evaluation of evidence, and coherent mechanistic synthesis across disciplines, thereby supporting a comprehensive understanding of the shared neurophysiological substrates underlying the comorbidity between sleep disorders and depression.
Human vs. animal
3.3
In selecting literature from the database, both animal models and human clinical studies were systematically considered. Animal research provides crucial mechanistic insight into how sleep disturbances contribute to depression, allowing precise manipulation of genetic, environmental, and neurochemical factors to probe causal links among altered sleep architecture, circadian dysregulation, neuroinflammation, and depression-like behaviors (McEwen et al., 2015; Nollet et al., 2020). Such models facilitate controlled pharmacological or environmental interventions but cannot fully capture the heterogeneity, subjectivity, and species-specific sleep–circadian features of human depressive disorders, which limits translational applicability (LeGates et al., 2014; Nestler and Hyman, 2010).
In contrast, human epidemiological, longitudinal, and clinical studies provide direct ecological and clinical validity, documenting real-world insomnia, hypersomnia, circadian phase delay, and irregular rest–activity rhythms and their associations with depressive onset, severity, and treatment response (Baglioni et al., 2011; Fang et al., 2019). Technological advances, such as actigraphy, wearable monitoring, structured assessments, and multimodal functional measures, offer nuanced characterization of sleep–mood dynamics in daily life. Although these studies cannot experimentally manipulate underlying mechanisms, integrating clinical evidence with mechanistic insights from animal research is essential for linking preclinical hypotheses to clinically relevant models. Cross-species intermediate phenotypes and multimodal translational approaches thus remain central to clarifying how sleep and circadian disruption contribute to depression (Krause et al., 2017). Accordingly, this review treats human clinical research as primary evidence for shaping theoretical frameworks, with animal studies serving as complementary resources that enrich mechanistic understanding.
Sleep disorders
4
The etiology of sleep disorders is multifactorial, with insomnia being the most prevalent. This section examines contributing factors from two perspectives: lifestyle influences and comorbid medical conditions.
Lifestyle factors
4.1
Modern societal pressures, including chronic stress, alcohol consumption, and excessive internet use, are strongly associated with impaired sleep quality, typically manifesting as insomnia or insufficient sleep.
Chronic Stress: Chronic stress has emerged as a significant factor associated with sleep disturbances through both physiological and cognitive pathways. Empirical evidence indicates that stress not only correlates with objective sleep disruptions but also amplifies negative subjective perceptions of sleep via cognitive distortions (Alfasi and Soffer-Dudek, 2018). For instance, prolonged sleep latency, early awakenings, and parasomnias have been associated with a reduction in total sleep time of approximately 10 % (Otsuka et al., 2017). This bidirectional relationship may create a self-reinforcing cycle of stress, sleep disturbance, and psychological distress. Dysregulation of endocrine function (e.g., altered melatonin secretion) and maladaptive cognitive appraisal highlight the potential value of stress-reduction strategies in sleep health interventions. Longitudinal and interventional studies are warranted to confirm causal relationships.
Alcohol Consumption: Alcohol exerts complex effects on sleep. Earlier research suggested that moderate alcohol intake before bedtime might facilitate sleep onset (Johnson et al., 1998); however, subsequent studies have demonstrated that alcohol disrupts normal sleep architecture, producing altered sleep patterns. Consequently, alcohol is now considered a potentially detrimental sleep aid (Su et al., 2010). Acute intake may shorten sleep latency but impairs overall sleep quality, likely through modulation of cortical GABAergic systems and dopaminergic pathways (Koob and Colrain, 2020). Chronic use and subsequent abstinence-related sleep disturbances are associated with GABAA receptor desensitization, impaired dopaminergic signaling, hyperactivation of stress modulators (e.g., orexin, norepinephrine, CRF, and pro-inflammatory cytokines), and glutamatergic dysregulation. Future longitudinal studies are needed to clarify the temporal and causal dynamics.
Internet Addiction: Classified as a behavioral disorder, internet addiction is strongly associated with psychiatric conditions and sleep disturbances (Zhang et al., 2017). Excessive engagement with digital devices, including smartphones, computers, and televisions, may exacerbate stress and anxiety, contributing to sleep problems in both children and adults (Alfasi and Soffer-Dudek, 2018). Specific online activities, such as messaging and emailing, have been correlated with chronic stress and depressive symptoms, further increasing the risk of sleep disturbances (Thomée et al., 2011). Interventional studies are required to determine whether reducing internet use can causally improve sleep outcomes.
Comorbid medical conditions
4.2
Sleep disturbances associated with medical conditions are often less modifiable than those related to lifestyle factors. These include sleep impairments secondary to neurodegenerative diseases or other physiological conditions, as well as insomnia or narcolepsy triggered by disease episodes.
Neurodegenerative Diseases: Parkinson's disease (PD), a prototypical neurodegenerative disorder, is frequently associated with disruptions in sleep-wake regulation (Zuzuárregui and During, 2020). Pathological hallmarks involve progressive degeneration of neural circuits that regulate sleep-wake cycles, affecting neurotransmitter systems involved in sleep modulation, including noradrenergic, serotonergic, dopaminergic, and GABAergic pathways (Samizadeh et al., 2023). Imbalances in these systems have been linked to sleep disturbances, with common clinical manifestations including sleep fragmentation and reduced restorative sleep, characteristic features of insomnia (Chahine et al., 2017). Longitudinal studies are necessary to determine causal links between specific neurotransmitter dysregulation and sleep outcomes.
Other Physiological Factors: Chronic pain and nocturia are recognized as predisposing factors for sleep disturbances (Ito et al., 2020; Radziunas et al., 2018). These conditions disrupt sleep continuity through forced nocturnal arousals, leading to cumulative sleep deprivation and reduced sleep quality.
Neurophysiological mechanisms
4.3
Overall, dysregulation of sleep-regulating neurotransmitters and hormones, such as serotonin (Ito et al., 2020), melatonin (Prodhan et al., 2021), and dopamine (Samizadeh et al., 2023), represents a core neurophysiological basis for sleep disorders. While multiple factors contribute to sleep disturbances, neurological dysfunction appears central. Future interventional and longitudinal studies are essential to clarify causality and to develop targeted therapeutic strategies.
Depression
5
Etiology and risk factors
5.1
Environmental factors
5.1.1
The pathogenesis of depression is widely recognized as involving a complex interplay between genetic predisposition and environmental influences. From an environmental perspective, chronic stress, early-life traumatic experiences (including physical abuse and emotional neglect), and dysfunctional family dynamics have been consistently associated with increased vulnerability to depression. These exposures are thought to interact with genetic background through mechanisms such as epigenetic modifications, thereby contributing to heightened risk of depressive disorders (Heim and Binder, 2012). The gene–environment interaction framework thus provides a useful theoretical basis for understanding the multifactorial etiology of depression.
Heim and Binder (2012) demonstrated that depression reflects the interaction of genetic susceptibility—including polymorphisms, familial psychiatric history, neuroendocrine characteristics, and personality traits—with adverse childhood experiences. Such pathogenic exposures are linked to enduring epigenetic alterations and sustained neurobiological dysregulation, including HPA axis hyperactivity, neurotransmitter imbalances, and structural as well as functional alterations in neural circuits. These maladaptive changes appear to increase susceptibility to depression and may also predispose individuals to comorbid medical conditions. Early interventions and comprehensive, multidisciplinary preventive approaches are therefore considered essential for reducing both individual suffering and the societal burden of depression. Longitudinal research is required to clarify the causal pathways linking specific environmental exposures to the onset of depressive disorders.
Neurogenic basis
5.1.2
Recent evidence has increasingly emphasized the neurogenic basis of depression, expanding upon the classical monoamine hypothesis toward a neurotrophic–neurogenic model. This hypothesis posits that impaired adult hippocampal neurogenesis contributes to the pathophysiology of depression, particularly under chronic stress conditions. Eisch and Petrik (2012) reported that reduced proliferation and differentiation of neural progenitor cells in the dentate gyrus are associated with depressive-like behaviors, and that improvement of these deficits through antidepressant treatment is critical for symptom alleviation. Similarly, Lucassen et al. (2010) found that stress-induced dysregulation of the HPA axis is associated with impaired adult neurogenesis, correlating with depressive phenotypes.
Neurogenic biomarkers—including brain-derived neurotrophic factor (BDNF), Ki-67, doublecortin (DCX), and hippocampal volume—have shown consistent associations with depression severity. For example, Karege et al. (2002) observed significantly reduced serum BDNF levels in depressed patients compared with controls, with levels increasing after pharmacological treatment in parallel with reductions in Hamilton Depression Rating Scale (HAMD) scores. Structural MRI studies further demonstrate that reduced hippocampal volume, particularly in subfields such as CA1, CA3, and the dentate gyrus, is negatively correlated with depressive symptom severity (Huang et al., 2013). Mechanistically, BDNF signaling has been shown to promote neuroplasticity and stress resilience, suggesting that restoration of neurogenesis is both a mediator and an outcome of effective antidepressant treatment (Duman and Monteggia, 2006). Taken together, these findings support the potential of neurogenesis-related markers as both diagnostic indicators and therapeutic targets. Future interventional and longitudinal studies are necessary to determine whether impaired neurogenesis plays a causal role in depression or represents an associated consequence.
Neurobiological mechanisms
5.2
The pathophysiology of depression encompasses multiple hypotheses (Li et al., 2021), including: Monoamine hypothesis (Dysregulation of serotonin, norepinephrine, and dopamine); HPA axis dysfunction (Hyperactivity in stress-response pathways); Glutamatergic signaling (Excitotoxicity and synaptic plasticity deficits); GABAergic inhibition (Reduced GABA neurotransmission); Neurotrophic factors: (Impaired brain-derived neurotrophic factor signaling); Neuroinflammation (Elevated pro-inflammatory cytokines).
Furthermore, substantial evidence demonstrates mechanistic links between depression and sleep disorders. For example, Petit et al. (2021) identified significant associations between glucose metabolic dysregulation and both depressive symptoms and sleep regulation. On the other hand, Yang et al. (2023) revealed elevated levels of inflammatory biomarkers in individuals with either depression or insomnia, which shew that there may be shared pathophysiological pathways. Overall, these multidisciplinary studies demonstrate that sleep disorders and depression are not independent, unrelated conditions, but rather exhibit significant intersecting and overlapping relationships.
Neurophysiological analysis
6
Key neurobiological findings with effect size estimates
6.1
Table 1 summarizes the key neurobiological characteristics of sleep–depression comorbidity using standardized effect size metrics, providing multidimensional interpretive value. Reduced cortical GABA levels (d = −0.73) and elevated evening cortisol (OR = 1.52) demonstrate moderate-to-large effect sizes, suggesting GABAergic dysfunction and HPA axis hyperactivity as central features. Conversely, null findings for BDNF (p > 0.05) highlight limitations of the neurotrophic hypothesis in explaining comorbidity. Cross-system comparisons reveal that GABA dysregulation (d = −0.73) exerts a larger effect than inflammatory marker IL-8 (β = 0.30), indicating potential hierarchical contributions across neurobiological systems. These quantitative data provide an objective framework for understanding pathophysiology, inform clinical prioritization of GABA modulation and HPA axis monitoring, and underscore the need for future studies to examine cross-system interactions in greater detail.Table 1. Key neurobiological findings with effect size estimates.Table 1. BiologicalSystemMeasureEffectSize(95 %CI)p-valueStudyGABAergicCortical GABAd = −0.73 (−1.12, −0.34)<0.001Benson et al. (2020)HPA AxisEvening cortisolOR = 1.52 (1.12, 2.06)0.008Pires Santiago et al. (2020)InflammationIL-8 levelsβ = 0.30 (0.15, 0.45)0.001Yang et al. (2023)NeuroplasticityBDNFNS (p > 0.05)–Pires Santiago et al. (2020)
Pathological association mechanisms
6.2
Through analyses of clinical symptomatology and pathogenic mechanisms, a significant pathological association emerges between sleep disorders and depression. Neuroimaging and biochemical studies consistently support the role of neurophysiological mechanisms in the onset and progression of depression. Structural MRI investigations have demonstrated cortical thinning in prefrontal regions and reduced functional connectivity within the default mode network, while biochemical evidence highlights reduced serum BDNF levels, elevated cortisol concentrations, and alterations in REM latency as correlates of depression severity (Molendijk et al., 2014). Complementary EEG findings show a reduction in NREM stage 2 sleep duration (−22 min; p < 0.05) and shortened REM latency, reflecting HPA axis dysregulation and altered monoaminergic signaling (Plante, 2021; Wang et al., 2015).
At the therapeutic level, pharmacological evidence suggests a partial overlap in the pathogenic mechanisms of depression and sleep disorders. For instance, selective serotonin reuptake inhibitors (SSRIs) have been shown to alleviate both depressive symptoms and co-occurring sleep disturbances (Fava et al., 2006). As noted by Dhuna and Malkani (2020), these medications act primarily through monoaminergic neurotransmitter systems, which are integral to both affective regulation and the sleep–wake cycle. Emerging research continues to refine understanding of this neurobiological interdependence, emphasizing common neurotransmitter and neurohormonal substrates that link mood and sleep regulation.
Neuroimaging findings further substantiate these associations, revealing that patients with either insomnia or depression frequently exhibit similar neuroanatomical changes, including diminished cortical surface area in prefrontal regions (Plante, 2021). Such findings suggest that sleep disturbances may not simply represent secondary manifestations of depression but could serve as early neurobiological markers or prodromal features of mood disorders (Jackson et al., 2003). More granular analyses confirm significant cortical thinning in prefrontal areas among depressed individuals with sleep disturbances (mean reduction = 0.15 mm, 95 % CI: 0.12–0.18 mm, p < 0.001) (Sha et al., 2019). Parallel EEG investigations reveal consistent reductions in NREM stage 2 duration (mean = −22 min, SD = 5.2, p = 0.003) and shortened REM latency (20–30 % reduction, p < 0.01) (Plante, 2021). Nonetheless, these findings require cautious interpretation in light of inconsistent biomarker results. For example, Pires Santiago et al. (2020) reported non-significant BDNF changes, challenging straightforward neurotrophic explanations.
Taken together, current evidence suggests that sleep disorders and depression are linked by bidirectional pathophysiological mechanisms, with shared neurobiological substrates that extend beyond simple symptomatic overlap. Beyond these pathological correlations, substantial evidence implicates multiple neurochemical factors—including neurotransmitters and hormones—in the co-occurrence of sleep disturbances and depression, as systematically reviewed below.
Neurotransmitter systems
6.3
Neurotransmitters play a central role in the pathogenesis of both depression and primary insomnia, a relationship schematically illustrated in Fig. 2. Among these systems, GABA, serotonin (5-HT), dopamine, and norepinephrine are most consistently implicated. Dysfunction within these neurotransmitter networks provides a neurochemical basis for the frequent co-occurrence of depressive disorders and sleep disturbances.
GABAergic dysfunction has been strongly linked to both depression and sleep disorders (Benson et al., 2020). Reduced GABAergic activity has been observed in patients with insomnia and major depressive disorder, and sleep disturbances may further aggravate depressive symptoms by lowering central GABA levels. Conversely, depression can impair sleep regulation through overactivation of the HPA axis, which suppresses GABAergic inhibition (Hepsomali et al., 2020; Li et al., 2021; Luscher et al., 2011). This reciprocal interaction forms a self-perpetuating cycle in which sleep disruption and affective dysregulation reinforce one another.
Beyond GABA, serotonin and dopamine play critical roles in the regulation of wakefulness, sleep architecture, and affective stability. Serotonin exerts dual effects: while reduced serotonergic activity has been consistently associated with more severe depressive symptoms and higher suicidality risk (Luscher et al., 2011), excessive serotonergic signaling can increase REM sleep expression while disrupting sleep continuity, thereby degrading sleep quality (Alexandre et al., 2006; Monti and Jantos, 2008). Dopamine, by contrast, is central to reward and motivation circuits. Abnormal dopaminergic transmission has been linked to insomnia and depressive symptoms, in part through a feedforward loop that exacerbates dopamine system dysfunction (Finan and Smith, 2013). Such imbalances ultimately compromise both mood regulation and sleep stability.
Norepinephrine, another key monoaminergic neurotransmitter, exerts dual regulatory effects on emotional processing and sleep–wake control. Pharmacological evidence shows that monoamine reuptake inhibitors, which elevate extracellular norepinephrine and serotonin, suppress REM sleep while alleviating depressive symptoms (Wilson and Argyropoulos, 2005). However, the link between REM sleep abnormalities and depression is not entirely linear. More recent studies suggest that while monoaminergic systems regulate both REM sleep and affective states, the underlying neural pathways may be distinct but interconnected (Wang et al., 2012).
Taken together, these findings indicate that disturbances across GABAergic and monoaminergic systems contribute to both depression and primary insomnia. Neurotransmitter imbalances not only impair sleep quality but also destabilize mood, highlighting the shared neurochemical substrates that underpin their bidirectional relationship.
Hormonal regulation
6.4
Key hormonal factors, notably cortisol and melatonin, have been identified as significant biomarkers implicated in both sleep disorders and depression. These neuroendocrine markers have attracted considerable attention for their potential involvement in the shared pathophysiology of the two conditions. Fig. 3 illustrates this relationship: both disorders are associated with excessive activation of the HPA axis, abnormal cortisol secretion, and the emergence of a reinforcing cycle that links them. The following sections provide a detailed explanation.
Neurobiological studies suggest that hyperactivation of central cholinergic neurons may constitute a common neurobiological substrate for sleep disorders (e.g., shortened REM sleep latency) and depressive symptoms (e.g., dysregulated cortisol secretion rhythms) in individuals with depression (Poland et al., 1989). This dysfunction indicates that disruptions in cholinergic signaling are associated with downstream neuroendocrine imbalances, which may contribute to the amplification of pathological processes.
Subsequent research has highlighted the pivotal role of the HPA axis in both sleep–wake regulation and emotional modulation. Cortisol, a key stress hormone, plays a critical role in sleep onset and circadian rhythm maintenance. Dysregulation of the HPA axis is frequently observed in individuals with depression, and the severity of this dysregulation has been reported to correlate with stress exposure, depressive symptom intensity, and illness duration (Pires Santiago et al., 2020). These findings suggest that HPA axis dysfunction and abnormal cortisol secretion patterns may represent a central physiological link between sleep disturbances and major depressive disorder.
Melatonin, commonly referred to as the “sleep hormone,” not only regulates circadian rhythms but also contributes to the maintenance of healthy sleep patterns. Disruptions or reductions in melatonin secretion have been reported in individuals with depression. Consequently, therapeutic strategies such as light therapy, sleep deprivation therapy, and melatonin supplementation have been investigated for their potential to improve both depressive symptoms and sleep disturbances (Germain & Kupfer, 2008; Parry et al., 2019).
The immune-inflammatory system
6.5
A growing body of evidence indicates that aberrant activation of the immune–inflammatory system plays a central role in the pathophysiology of depression, while sleep disturbances further amplify depression risk via inflammatory pathways (Veler, 2023). Depressed individuals frequently exhibit elevated peripheral pro-inflammatory markers, including interleukin-1β (IL-1β), interleukin-6 (IL-6), tumor necrosis factor-α (TNF-α), and C-reactive protein (CRP) (Yang et al., 2023). These inflammatory signals can affect brain function through blood–brain barrier permeability, vagal afferent signaling, or microglial activation, inducing affective and cognitive changes characteristic of sickness behavior (Beurel et al., 2020; Dantzer et al., 2008). Sleep disturbances, particularly insomnia, show a robust bidirectional relationship with inflammation: sleep loss promotes systemic inflammatory activation, whereas inflammatory states disrupt sleep architecture and homeostasis, forming a self-reinforcing cycle (Irwin, 2019).
At the molecular level, pro-inflammatory cytokines (e.g., IL-6, TNF-α, IFN-γ) activate indoleamine 2,3-dioxygenase (IDO), diverting tryptophan metabolism from serotonin synthesis toward the kynurenine pathway, thereby reducing central serotonin availability and generating neuroactive metabolites such as quinolinic acid (Dantzer et al., 2008; Miller et al., 2009). In parallel, inflammation-induced microglial activation impairs synaptic integrity and neuroplasticity and disrupts circadian regulation via effects on the suprachiasmatic nucleus and melatonin rhythms (Irwin and Opp, 2017; Yirmiya et al., 2015). As Fig. 4 shown, together, these mechanisms provide a biological framework for depression–sleep disorder comorbidity and support integrated interventions targeting both inflammation and sleep dysfunction.
Additional mechanisms
6.6
Emerging evidence implicates the glutamate (Glu)/GABA system, and astrocytic metabolism in the pathophysiology of both sleep disorders and depression.
The Glu/GABA system plays a fundamental role in regulating both mood and sleep. Glutamate functions as the brain's primary excitatory neurotransmitter, while GABA—synthesized from glutamate—serves as the main inhibitory counterpart (Zhang et al., 2023). This excitatory–inhibitory balance is critical for normal brain function. In depression, evidence indicates that this balance is frequently disrupted, with excitatory and inhibitory signaling becoming misaligned (Chaves et al., 2018). In insomnia, shorter sleep duration has been hypothesized to associate with reduced glutamate levels (Benson et al., 2020). As depicted in Fig. 5, dysregulation of the Glu/GABA system likely represents a shared pathophysiological mechanism underlying both depression and sleep disorders.
Recent advances in neuroglial research have also highlighted the role of astrocytes in linking metabolism with neuropsychiatric conditions. Specific astrocytic subtypes regulate glucose metabolism in the central nervous system, contributing to neuroprotection, gliotransmission, and homeostatic stability (MacMahon Copas et al., 2021). Astrocytes are the primary sites for GABA uptake and play a key role in its synthesis (Zhang et al., 2023). Dysfunction in these astrocytic processes can disrupt brain energy balance and has been linked with mood and sleep disturbances (J.-M. Petit et al., 2021). As illustrated in Fig. 6, astrocytes influence these disorders indirectly, primarily through their regulation of neurotransmitter metabolism and neural energy supply.
Discussion
7
Overview
7.1
Sleep disturbances and depression are highly comorbid, with growing evidence suggesting a complex, bidirectional relationship. Rather than representing independent clinical phenomena, recent findings indicate that these conditions may share overlapping neurobiological substrates. Based on the aforementioned common theories and hypotheses, the neurobiological link between sleep disorders and depression is illustrated in Fig. 1.Fig. 1A simplified diagram of neurobiological substrates involved in sleep disorders and depression.Fig. 1
Neuroimaging and electrophysiological studies consistently support this association. Structural MRI investigations have reported cortical thinning and reduced surface area in the prefrontal and anterior cingulate cortices among individuals with depression—regions also implicated in insomnia and impaired emotion regulation (Sha et al., 2019). Functional connectivity analyses further reveal disruptions within the default mode and salience networks across both disorders. Complementary EEG data demonstrate reduced non-REM stage 2 sleep duration (approximately −22 min; p < 0.05) and shortened REM sleep latency in depressed individuals, alterations linked to hyperactivation of the HPA axis and monoaminergic dysregulation (Plante, 2021).
Overlap in neurotransmitter dysfunction provides additional evidence of shared mechanisms. GABAergic deficits have been implicated in both insomnia and depression, with magnetic resonance spectroscopy studies reporting lower cortical GABA concentrations (Benson et al., 2020; Hepsomali et al., 2020). Antidepressants such as SSRIs have been shown to partially restore GABAergic tone while concurrently improving depressive and sleep-related symptoms (Nikhil A. Dhuna & Roneil G. Malkani, 2020). 5-HT, while essential for sleep initiation and mood stabilization, may also disrupt sleep continuity when present in excess, leading to increased REM density (Monti and Monti, 2007). Dysregulation of dopamine and norepinephrine further contributes to impaired reward processing and disrupted sleep–wake regulation. The HPA axis serves as a hormonal interface between sleep and affect, with elevated cortisol frequently observed in both acute insomnia and chronic depression. For instance, Pires Santiago et al. (2020) reported that the severity of HPA axis dysregulation correlates with illness duration and depressive symptom severity. Suppression of melatonin secretion, commonly observed in these conditions, has also motivated chronotherapeutic approaches such as bright light therapy and melatonin supplementation to address circadian and affective disturbances (Parry et al., 2019).
Beyond classical neurotransmitter models, emerging evidence highlights the contribution of inflammatory signaling, Glu/GABA imbalance, and astrocytic dysfunction to the co-occurrence of sleep disturbances and depression. Elevated pro-inflammatory cytokines, including IL-6 and TNF-α, have been consistently reported in individuals with poor sleep quality and depressive symptoms (Irwin and Opp, 2017). These inflammatory processes appear to influence both sleep architecture and mood regulation. Additionally, excitatory–inhibitory imbalance, characterized by altered glutamate and GABA transmission, has been associated with cortical hyperexcitability and emotional dysregulation (Chaves et al., 2018; Zhang et al., 2023). Astrocytes play a critical role in maintaining this balance by regulating glutamate clearance and GABA reuptake. Dysfunction in astrocytic metabolism has been linked to impaired energy homeostasis and excitatory–inhibitory imbalance, thereby increasing vulnerability to mood and sleep disorders (Hertz, 2004; J.-M. Petit et al., 2021). These findings collectively support a model in which overlapping neurochemical, hormonal, and glial mechanisms contribute to the frequent co-occurrence of insomnia and depression. However, longitudinal and interventional studies are required to confirm the causal pathways underlying these associations.
In summary, the co-occurrence of sleep disturbances and depression reflects multifactorial interactions encompassing structural and functional brain abnormalities, neurotransmitter imbalances, neuroendocrine dysregulation, and astrocytic dysfunction. These shared mechanisms underscore the importance of integrated diagnostic and therapeutic strategies that address mood and sleep disturbances simultaneously. Future research should emphasize longitudinal, mechanistic, and interventional designs to clarify causal pathways and inform precision medicine approaches.
Limitations
7.2
Despite the breadth and integrative scope of this review, several limitations warrant consideration.
- (a)Etiological ambiguity. Extensive literature review reveals that, persistent epistemological and methodological limitations in contemporary research on the etiology of sleep disorders, which remains largely dominated by epidemiological designs and self-reported measures. Although such approaches have been valuable for identifying associations and population-level risk factors, they are inherently constrained to descriptive and correlational inference and offer limited insight into the neurobiological mechanisms that generate heterogeneous sleep pathologies. Insomnia, in particular, is frequently conceptualized as a unitary clinical entity, yet accumulating evidence suggests marked etiological heterogeneity: similar phenotypic manifestations may arise from fundamentally distinct neurophysiological processes. For example, delayed sleep onset resulting from circadian misalignment induced by artificial light exposure is unlikely to share identical mechanisms with sleep disruption secondary to systemic medical or inflammatory conditions, despite superficial clinical similarity. These pathways may differentially engage the HPA axis, melatonergic signaling, autonomic regulation, and affective neural circuits, underscoring the risk of mechanistic oversimplification when phenotypes are treated as homogeneous. Etiological ambiguity is even more pronounced in mood disorders such as depression, where sleep disturbance is both a core symptom and a prognostic factor. Although gene–environment interaction models dominate the field, they have yet to yield definitive causal accounts. Dysregulation of serotonergic transmission and sustained HPA axis hyperactivity are consistently observed in depressive states, but their causal status remains unresolved: it is unclear whether these alterations initiate depressive pathology or emerge as secondary adaptations to chronic stress and disturbed sleep. This uncertainty has substantial translational consequences. First-line pharmacotherapies, particularly selective serotonin reuptake inhibitors, largely provide symptomatic relief without clearly rectifying upstream biological perturbations, potentially explaining their variable efficacy and delayed onset of action. Addressing this gap requires a conceptual shift toward disentangling primary pathogenic mechanisms from compensatory neurochemical and neuroendocrine responses.
- (b)Publication Bias. The current evidence base is highly vulnerable to publication bias. Positive findings demonstrating significant associations between sleep disturbances and depressive outcomes are disproportionately represented in the published literature, whereas null or negative studies are less likely to be reported. This bias may artificially inflate the strength of observed associations and foster premature causal inferences. Meta-analyses of insomnia and depression consistently show asymmetry suggestive of selective reporting (Baglioni et al., 2011; Doi et al., 2022). Future reviews should implement formal statistical assessments of publication bias (e.g., funnel plots, Egger's regression) and include unpublished or gray literature to mitigate overestimation of effect sizes.
- (c)Population Heterogeneity. Most included studies inadequately account for demographic and sociocultural heterogeneity. Age, sex, cultural background, and socioeconomic status all influence both the prevalence and neurophysiological mechanisms of sleep–depression comorbidity. For instance, adolescence and late adulthood are peak vulnerability periods (Malhi et al., 2018), while sex differences in hormonal transitions (e.g., perinatal or menopausal states) strongly modulate risk (Parry et al., 2019). Moreover, cultural determinants such as chronotype variability and societal stress exposure shape both sleep and mood outcomes (Roenneberg and Merrow, 2016). The limited consideration of such moderators constrains generalizability and risks obscuring subgroup-specific pathways. Addressing these gaps will require stratified analyses, meta-regressions incorporating demographic moderators, and prospective multicenter cohorts across cultural contexts.
- (d)Translational Gap. Another limitation concerns translational validity. Much molecular and neurophysiological evidence derives from animal models, particularly rodent paradigms of stress-induced insomnia or depressive-like behavior. While such models yield critical insights into HPA axis dysregulation and glutamatergic imbalance, extrapolation to humans is inherently constrained due to species differences in sleep architecture, immune signaling, and higher-order cognition (Kendler, 2019; Nestler and Hyman, 2010). Moreover, rodent paradigms often emphasize acute stress, failing to capture the chronic, multifactorial nature of human comorbidity. Bridging this gap requires more robust human studies, including longitudinal biomarker designs and clinical trials integrating mechanistic endpoints, to validate animal-derived hypotheses within clinically relevant contexts (Insel, 2014).
- (e)Insomnia Bias. Finally, research disproportionately emphasizes insomnia relative to other sleep disorders. Although insomnia is highly prevalent and a strong predictor of depression, this narrow focus risks constraining theoretical models and clinical translation. Disorders such as obstructive sleep apnea, circadian rhythm sleep–wake disorders, and restless legs syndrome also show robust associations with depression but remain underexamined (Kendzerska et al., 2014; Winkelman et al., 2008). This insomnia-centric bias limits external validity. Future research should broaden the scope to include diverse sleep disorders, enabling comparative analyses and refining mechanistic frameworks.
Future Directions for Addressing Limitations. To overcome these limitations, several strategies are recommended. First, prospective meta-analyses should adopt bias-detection tools and incorporate gray literature to reduce publication bias. Second, primary studies should stratify analyses by age, sex, and socioeconomic status, and conduct cross-cultural investigations. Third, translational integration can be strengthened by aligning preclinical models with clinical endpoints and employing reverse-translation frameworks. Finally, expanding beyond insomnia through multicenter, cross-diagnostic cohorts will enhance generalizability. Together, these strategies will improve methodological rigor, strengthen causal inference, and guide more personalized and culturally sensitive interventions.
Proposed future research directions
7.3
Future research should prioritize large-scale, multicenter prospective cohort studies (target n > 500) with standardized multimodal assessments at baseline and follow-ups at 1 and 3 years. These assessments should include polysomnography (PSG), structural and functional MRI (high-resolution structural, resting-state connectivity, and optional task-based paradigms), and repeated serum and, when ethically permissible, cerebrospinal fluid sampling. Biomarkers should encompass HPA axis activity (free cortisol), inflammatory markers (IL-6, TNF-α, CRP), neurotrophic factors (BDNF), and indices of excitatory–inhibitory balance (MRS or plasma measures of Glu/Gln and GABA), complemented by astrocytic markers such as GFAP, S100B, and glycogen metabolism enzymes. Cohorts should systematically collect and adjust for major confounders (age, sex, BMI, lifestyle factors, comorbidities, and psychotropic/anti-inflammatory medication) and employ pre-registered analytic strategies with rigorous correction for multiple comparisons and transparent handling of missing data. Such multimodal, longitudinal designs are warranted by consistent epidemiological evidence linking sleep disturbances to increased risk of depressive episodes and allow mechanistic dissection of sleep–immune–neurotrophic–astrocytic pathways (Baglioni et al., 2011; Li et al., 2016; Wulff et al., 2010).
In parallel, stratified randomized controlled trials (RCTs) should be conducted to test both mechanistic and therapeutic hypotheses. Pharmacological mechanism trials should evaluate candidate compounds with astrocytic or metabolic targets (e.g., modulators of glycogenolysis, purinergic signaling, or glutamate clearance), with biomarker-based stratification and outcomes encompassing PSG-derived sleep continuity, sleep quality, depression severity (MADRS/HAM-D), inflammatory cytokines, BDNF, and imaging changes. Adjunctive sleep-targeted interventions (e.g., hypnotics or CBT-I combined with antidepressants) should be compared against antidepressant-only strategies in preregistered, blinded designs, with remission speed and stability as primary endpoints and biomarker shifts as secondary measures. To maximize reproducibility, standardized PSG, biospecimen, and MRI protocols are required, with resulting multimodal datasets enabling machine learning and causal inference approaches supported by external and cross-cultural validation. Data and biospecimen sharing under ethical frameworks will accelerate translation into risk predictors and companion diagnostics. Finally, given the temporal dynamics of sleep–mood interactions, future work should incorporate population-specific and chronotherapeutic approaches (e.g., timed wake therapy, light exposure) across developmental stages to identify individualized therapeutic windows, consistent with emerging evidence on sleep–inflammation mechanisms and chronotherapy (Depner et al., 2014; Irwin, 2019; Singh et al., 2021; Verkhratsky et al., 2021; Wu et al., 2015).
Conclusion
8
The intricate bidirectional interaction between sleep disorders and depression reflects a shared neurophysiological substrate involving neurotransmitter dysregulation, hormonal and circadian rhythm disruption, as well as neuroinflammatory processes. These converging mechanisms not only account for the high degree of comorbidity and symptom overlap between the two conditions but also offer promising targets for integrated therapeutic strategies. For example, these findings reinforce the hypothesis of shared neurobiological substrates in sleep disorders and depression. EEG and neuroimaging data reveal overlapping cortical abnormalities and HPA axis dysfunction. Pharmacological responses to SSRIs and melatonin-based therapies further confirm the co-involvement of monoaminergic and circadian systems. Nonetheless, the directionality remains underexplored. This framework advocates for a paradigm shift beyond traditional diagnostic boundaries, embracing a holistic approach to the treatment of comorbid psychiatric disorders. Progress in this field will require interdisciplinary collaboration and longitudinal, cross-cultural research designs to inform the development of more effective, personalized interventions that address dysfunctions in both sleep disorders and depression.
Declaration of competing interest
I have nothing to declare.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Alexandre C.Popa D.Fabre V.Bouali S.Venault P.Lesch K.-P.Hamon M.Adrien J.Early life blockade of 5-Hydroxytryptamine 1A receptors normalizes sleep and depression-like behavior in adult knock-out mice lacking the serotonin transporter J. Neurosci.262020065554556410.1523/jneurosci.5156-05.200616707806 PMC 6675294 · doi ↗ · pubmed ↗
- 2Alfasi D.Soffer-Dudek N.Does alexithymia moderate the relation between stress and general sleep experiences?Pers. Indiv. Differ.1222018879210.1016/j.paid.2017.10.014 · doi ↗
- 3Al-Khalil Z.M.El Sheikh Lababidi G.H.Shehayeb E.O.Ghanime P.M.Talih F.R.Tamim H.Kaafarani B.R.Impact of socioeconomic and political stressors on mental health: a cross-sectional study on university students in Lebanon BMC Med. Educ.25120259110.1186/s 12909-025-06701-139827125 PMC 11743072 · doi ↗ · pubmed ↗
- 4Amagai Y.Ishikawa S.Gotoh T.Kayaba K.Nakamura Y.Kajii E.Sleep duration and incidence of cardiovascular events in a Japanese population: the Jichi medical school Cohort study J. Epidemiol.202201010611010.2188/jea.JE 2009005320009370 PMC 3900808 · doi ↗ · pubmed ↗
- 5Anacker C.Hen R.Adult hippocampal neurogenesis and cognitive flexibility — linking memory and mood Nat. Rev. Neurosci.186201733534610.1038/nrn.2017.4528469276 PMC 6261347 · doi ↗ · pubmed ↗
- 6Baattaiah B.A.Alharbi M.D.Babteen N.M.Al-Maqbool H.M.Babgi F.A.Albatati A.A.The relationship between fatigue, sleep quality, resilience, and the risk of postpartum depression: an emphasis on maternal mental health Bmc Psychol.11120231010.1186/s 40359-023-01043-336635743 PMC 9836926 · doi ↗ · pubmed ↗
- 7Baglioni C.Battagliese G.Feige B.Spiegelhalder K.Nissen C.Voderholzer U.Lombardo C.Riemann D.Insomnia as a predictor of depression: a meta-analytic evaluation of longitudinal epidemiological studies J. Affect. Disord.1351–32011101910.1016/j.jad.2011.01.01121300408 · doi ↗ · pubmed ↗
- 8Baldini V.Gnazzo M.Maragno M.Biagetti R.Stefanini C.Canulli F.Varallo G.Donati C.Neri G.Fiorillo A.Plazzi G.Suicidal risk among adolescent psychiatric inpatients: the role of insomnia, depression, and social-personal factors Eur. Psychiatr.6812025 e 4210.1192/j.eurpsy.2025.29PMC 1204172440015959 · doi ↗ · pubmed ↗