Temporal dynamics of ferroptosis markers and neuroprotective intervention in cerebral ischemia-reperfusion injury: Insights for therapeutic strategy development
Shuangyan Bao, Chaojie Liu, Qian Wang Wang, Chenggui Zhang, Hairong Zhao

TL;DR
This study explores how ferroptosis changes over time after a stroke and suggests that targeting ferroptosis could improve brain recovery.
Contribution
The study reveals the temporal dynamics of ferroptosis markers in cerebral ischemia-reperfusion injury and identifies a potential therapeutic window.
Findings
BBB disruption and altered GSH, Fe(2+), and MDA levels were observed after MCAO/R.
Ferroptosis markers like NRF2, xCT, and GPX4 were down-regulated, while HO-1 showed an inverse trend.
Ferrostatin-1 pretreatment protected against CIRI, unlike Erastin.
Abstract
This study examines the temporal dynamics of ferroptosis following cerebral ischemia-reperfusion injury (CIRI) to establish a theoretical framework for innovative therapies that enhance neuronal survival by mitigating ferroptosis. An experimental CIRI model was established in mice via middle cerebral artery occlusion and reperfusion (MCAO/R). Behavioral assessments were conducted, and blood-brain barrier (BBB) integrity was evaluated using transmission electron microscopy. Immunoblotting and ELISA were performed to determine ferroptosis dynamics post-MCAO/R. Additionally, CIRI mice received intraperitoneal injections of Ferrostain-1 (10 mg/kg/d) and Erastin (30 mg/kg/d). The effects of ferroptosis on CIRI were further verified through 2,3,5-Triphenyltetrazolium chloride and hematoxylin-eosin staining. MCAO/R induced BBB disruption, and was associated with a reduction in GSH activity…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
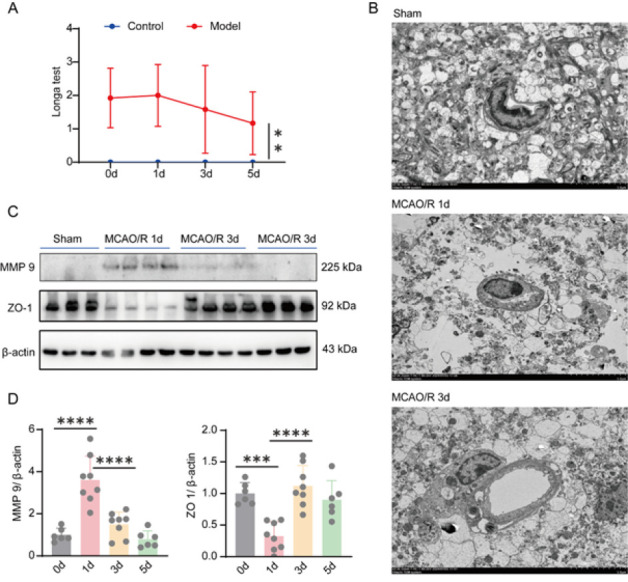 Figure 1
Figure 1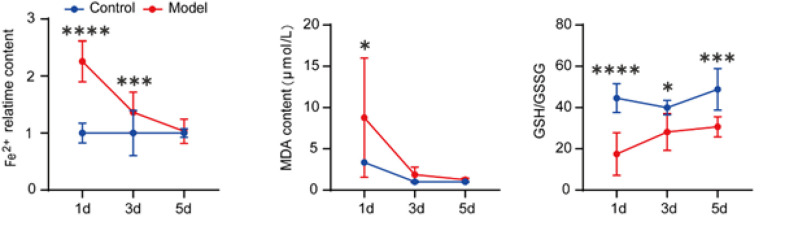 Figure 2
Figure 2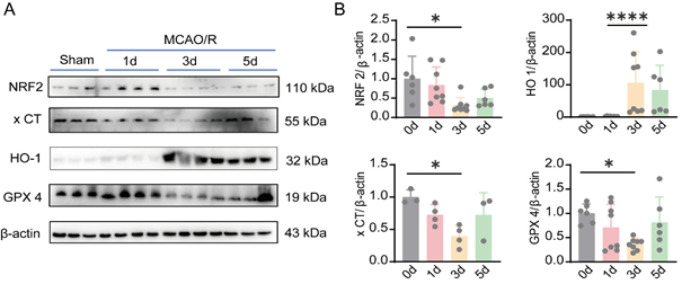 Figure 3
Figure 3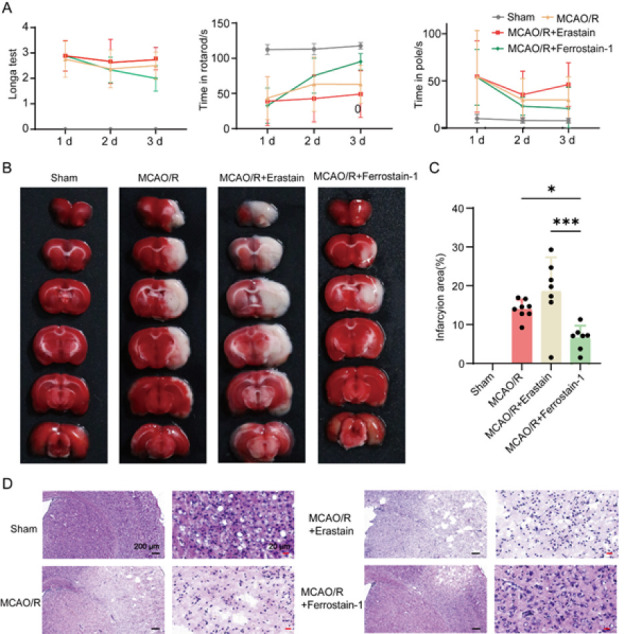 Figure 4
Figure 4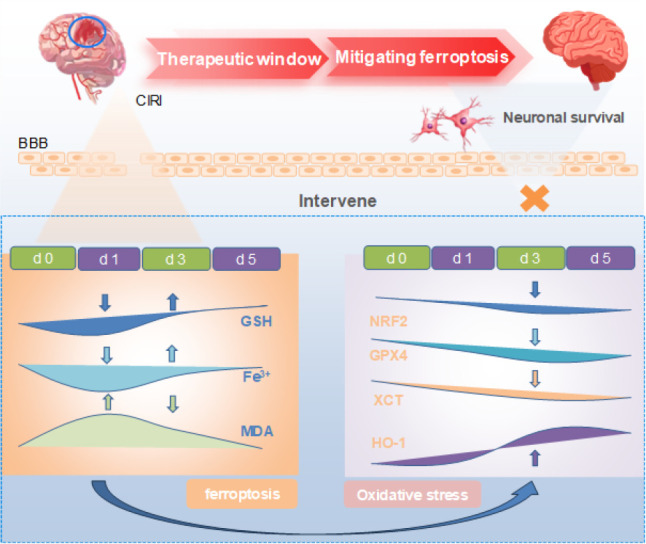 Figure 5
Figure 5Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsFerroptosis and cancer prognosis · Inflammation biomarkers and pathways · Cardiovascular Health and Risk Factors
Introduction
Stroke is a significant public health concern worldwide, as the third leading cause of death and the first leading cause of disability, which seriously affects human physical and mental health and quality of life (1, 2). A lack of blood flow causes ischemic stroke (IS); the incidence of IS is higher than that of hemorrhagic stroke, accounting for approximately 87% (3, 4). The pathogenesis of IS is very complex and is not fully understood (5, 6). CIRI and blood flow interruption after ischemia lead to hypoxia and glucose deficiency, energy metabolism disorders, oxidative stress, inflammatory responses, glutamate excitotoxicity, and other complications. Apoptosis, autophagy, necrosis, and pyroptosis can ultimately lead to neuronal death (7, 8). Various signaling pathways were activated in these pathological transitions, and their targeted regulation could serve as a potential therapeutic strategy. The pathophysiological process of IS is complex, and it is crucial to elucidate its accompanying damage and signaling mechanisms for the development of targeted interventions.
Dixon et al. were the first to describe ferroptosis in 2012 (9, 10). It is an iron-dependent cell death mechanism caused by the accumulation of lipid peroxides and related metabolites in the plasma membrane, as well as the depletion of polyunsaturated fatty acids (11, 12). Iron overload and a massive accumulation of lipid peroxides in cells define ferroptosis (12, 13). Cells accumulate iron after responding to stimuli. Excess iron ions eventually cause membrane lipid peroxidation via the Fenton reaction, resulting in mitochondrial damage, decreased function, and eventually, cell death (14, 15).
Ferroptosis is one of the mechanisms of ischemia-reperfusion injury, which is generally accompanied by ROS overproduction and iron accumulation after reperfusion. Excessive Fe (2+)-induced Fenton reaction (Fenton) produces hydroxyl radicals, which cause cytotoxicity (16, 17). Cerebral microvascular endothelial cell (BMVEC) dysfunction, ROS accumulation causes significantly increased macrophages, blood-brain barrier (BBB) damage, hemorrhagic transformation, and cerebral edema after cerebral infarction, and early neurological deterioration (18-20). Cerebral ischemia and reperfusion will cause brain damage, damage the integrity of the BBB, and sharply increase the iron content in the brain parenchyma, causing iron overload, lipid peroxidation, and other disorders of iron metabolism, which will eventually lead to cell iron death (21, 22). I/R leads to increased presynaptic glutamate release, decreased reuptake, increased free radical production, and promotes iron death. In 1990, Patt et al. conducted a study that indicated the involvement of iron in CIRI (23). Interestingly, brain iron has been linked to the severity of cerebral infarction, suggesting its role in ischemia-reperfusion injury (24). Furthermore, some studies have reported an association between iron overload-related death and CIRI; however, the dynamic monitoring of iron overload-related pathways after CIRI has not been investigated (19, 25). Therefore, we dynamically monitored the ferroptosis-related pathways after CIRI and clearly observed a dynamic process of ferroptosis, underscoring a therapeutic time window for ferroptosis-related CIRI. These insights provide a valuable theoretical foundation for developing novel CIRI treatment modalities.
Materials and Methods
Animals
Adult male C57BL/6 mice weighing 25-30 g (8-10 weeks) were supplied by the Dali University Laboratory Animal Center. They were habituated to a 12 hr light/dark cycle in a temperature-controlled environment (22-25 ^°^C) with a humidity of 40-70% and free access to food and water. All experimental procedures and animal housing in this study were approved by the Institutional Animal Care and Use Committee of Dali University, China (Animal ethics No.: DLU2017-0117).
Middle cerebral artery occlusion-reperfusion (MCAO/R) model
The MCAO procedure was performed according to the method outlined by Zea Longa, with improvements previously reported (26-28). The mice were anesthetized with an intraperitoneal injection of 1.25% tribromoethanol. All surgical materials and instruments were sterilized before use. The internal common carotid artery, external carotid artery (ECA), and internal carotid artery (ICA)(29, 30) were carefully separated, and a small incision was made in the CCA. Blood flow was obstructed using a sterile nylon suture (No.: MSMC21B120PK50, Ruiwode Bio-technology Co., Ltd., China) within the middle cerebral artery from the ECA to the ICA. After 60 min, the suture was removed to restore blood flow, thereby inducing MCAO/R. The sham group underwent the same procedures except that the suture was not inserted. During the surgery, the body temperature of the mouse was maintained at 36.5±0.5 ^°^C. The neurological score was assessed immediately after surgery, when the animal had fully recovered from anesthesia.
Behavior tests
The longa test, rotarod test, and pole test helped assess the neurological function of mice subjected to MCAO, according to our previously developed protocols. Rotarod test: The mice were placed on a rotating rod for the rotarod test (Shanghai Xinruan Information Technology Co., Ltd., China), with speeds incrementally increasing from 4 rpm to 40 rpm. The duration for each mouse to maintain its grip on the rotating rod was recorded (maximum time: 120 sec). Pole test: the mice were placed on the tip of an 8 mm (diameter) ×50 cm (length) wooden pole with its head upward. The mouse then attempts to descend to the floor by turning its head downward; meanwhile, the total time it takes to reach the floor is recorded.
TTC staining
The mice were equally divided into four groups: Sham, MCAO/R, MCAO/R+Erastain (30 mg/kg/day), and MCAO/R+Ferrostain-1 (10 mg/kg/day). Each group received daily treatment for three consecutive days. TTC staining was used to evaluate infarct size. The TTC solution was prepared in phosphate-buffered saline (PBS, pH 7.4) in the dark at 37 ^°^C immediately before use. For this assessment, the brain was rapidly isolated and sliced into 2-mm-thick coronal sections. Subsequently, the brain slices were stained with a 0.2% TTC solution at 37 ^°^C for 15 min and then fixed with 4% paraformaldehyde (PFA) for 24 hr. The infarct volume was analyzed using Image-Pro Plus version 6.0 image analysis software.
Transmission electron microscopy (TEM)
Mice were transcardially perfused with PBS, followed by the brain being sectioned into 1 mm-thick slices and post-fixed in 2.5% glutaraldehyde in PBS. Under a dissection microscope, tissue punches were taken to capture the IL and CL peri-infarct areas in the cortex. Tissues were washed three times in PBS, then post-fixed in 1% osmium tetroxide with 1% potassium ferricyanide for one hour. Following three additional PBS washes, the pellet was dehydrated through a graded series of 30-100% ethanol, 100% propylene oxide, and then infiltrated in a 1:1 mixture of propylene oxide: Polybed 812 epoxy resin for one hour. After several changes of 100% resin over 24 hrs, the pellet was embedded in a final change of resin, cured at 37 ^°^C overnight, followed by additional hardening at 65 ^°^C for two more days. Ultrathin (70 nm) sections were collected on 200 mesh copper grids, stained with 2% uranyl acetate in 50% methanol for ten minutes, followed by 1% lead citrate for seven minutes. Sections were imaged using a JEOL JEM 1011 transmission electron microscope (Peabody, MA) at 80 kV.
Determination of GSH/GSSG levels
The mice were deeply anaesthetized after 48 hr of I/R. The brains were collected in 1.5 ml EP tubes and homogenized with 500 µl 5% SSA (A610610-0050, DOJINGDO, Japan) solution. The samples were centrifuged at 8000× g for ten minutes, and the supernatant was transferred into a new tube. The supernatant was then diluted with ddH2O, and the concentration was adjusted to 0.5% with SSA. The concentration of GSH/GSSG was evaluated using the GSSG/GSH Quantification Kit II (G263, DOJINGDO, Japan). The absorbance was measured at 405 nm or 415 nm by Multiscan Spectrum (Multiskan Sky, Thermo, USA)
Brain MDA measurement
A brain sample of 10-30 mg was collected into 1.5 ml microtubes and homogenized with 300 µl of an antioxidant PBS solution over an ice bath. The mixture was then centrifuged at 10,000×g for five minutes. MDA content in brain tissues was analyzed using the MDA Assay Kit (M496, DOJINGDO, Japan). The samples were added 200 µl of Lysis Buffer and mixed thoroughly with a vortex oscillator, then left for five minutes at room temperature. The 300 µl working solution was added to each microtubule and mixed thoroughly with a vortex mixer. After heating in a constant temperature bath at 95 ^°^C for 15 min, the sample was cooled in an ice bath for five minutes and centrifuged at 10,000 ×g for ten minutes. 200 µl of supernatant was added to a transparent 96-well plate. The absorbance at 532 nm was measured using a Multiscan Spectrum. The MDA concentration in the sample was calculated by the standard MDA curve.
Iron assay
Additionally, the concentration of Fe (2+) in the mouse brain was determined using an Iron Assay Kit (I291, Dojindo Laboratories, Japan). Brain tissue was homogenized in cold PBS, and the supernatant was collected. The supernatant was then ultrasonicated for five minutes and centrifuged at 16,000×g for ten minutes to remove insoluble matter. The samples were divided into three parts: Fe (2+) detection, total Fe detection, and blank control. The sample for total Fe detection was supplemented with a reducing solution, and the remaining samples were supplemented with equal volumes of assay buffer. All samples were incubated for ten minutes at 37 ^°^C, then transferred into a 96-well plate and continued to incubate for an additional 60 min at 37 ^°^C. The iron content was measured using a Multiskan Spectrum (Multiskan Sky, Thermo, USA) at 593 nm.
Western blotting
The total protein was separated using RIPA lysis buffer with 1 mM PMSF (HY-L081, Med Chem Express, USA) and a protease inhibitor cocktail (HY-K0010, Med Chem Express, USA) for 15 min, followed by 15 min of centrifugation at 12000 rpm at 4 ^°^C. Protein concentration was measured using a BCA protein assay kit (P0011, Beyotime, China). Protein was separated with SDS-polyacrylamide gel electrophoresis, and then transferred from the gel onto a polyvinylidene difluoride (ISEQ00010, Millipore, USA) membrane. After being blocked with 5% nonfat dry milk in Tris-buffered saline plus Tween-20 (TBST) for 1.5 hr at room temperature, the PVDF membranes were incubated overnight at 4 ^°^C with primary antibodies, followed by 1 hour at room temperature with secondary antibodies. The membranes were washed with TBST buffer, and the blots were visualized by Azure C300. The information of antibodies is as follows: NRF2 (sc-7269, Santa Cruz Biotechnology), GPX4 (ab216327, abcom), XCT (ab38898, abcom, Goat anti-Rabbit 0295G-SA, Bioss, China, Goat anti-Mouse, 0296G-SA , Bioss, China) HO-1(10701-1-AP, Proteintech, USA).
HE staining
After collecting the tissues from each animal, the tissues were rinsed with water for two hours. After dehydration using different concentrations of ethanol, the tissues were dehydrated with xylene until they became transparent, embedded for one hour, and then sliced. Subsequently, the slides were roasted, dewaxed, hydrated, immersed in distilled water, and stained with an aqueous hematoxylin solution for three minutes, followed by differentiation with an ethanol-hydrochloric acid solution for 15 sec. After being slightly washed with water and blue returning solution for 15 sec, the slides were rinsed with water and stained with eosin for three minutes. Images were obtained using an inverted microscope (Zeiss, Oberkochen, Germany).
Statistical analysis
All data were presented as means and standard deviations. Means were compared using independent sample t-tests or one-way ANOVA. All experiments were performed in triplicate. A P-value of <0.05 indicated a significant difference.
Results
MCAO/R leads to disruption of the BBB
The BBB plays a crucial role as a key component of the neurovascular unit, and its impairment during brain ischemic injury can significantly exacerbate cerebral damage. In this study, we employed transmission electron microscopy to investigate the structural alterations occurring at the BBB in mice subjected to MCAO/R. First, as shown in Figure 1A, the behavioral score of mice after thread implantation was significantly different from that of normal mice, which suggests the successful establishment of the CIRI model. Immediately, the results of the TEM showed that, the induction of MCAO/R led to the disruption of basal lamina, tight junction proteins, and endothelial cells (ECs), as well as mitochondrial swelling and pronounced expansion of astrocytic end-feet (Figure 1B). This result suggests that the BBB of the mice was damaged after MCAO.
In addition, compared to the Sham group, a substantial up-regulation of MMP9 expression was observed on the initial day post-MCAO/R, followed by a subsequent decrease in its expression levels. Conversely, the expression levels of ZO-1 demonstrated an initial decline on the first day post-MCAO/R, succeeded by a subsequent increase (Figures 1C and D).
MCAO/R leads to a decrease in GSH and Fe (2+) secretion and an up-regulation of MDA
GSH/GSSH, MDA, and Fe (2+) serve as crucial biomarkers for assessing iron-induced cell death. This study scrutinized the variations in GSH/GSSH, MDA, and Fe (2+) levels in the brain tissues of mice at 1d, 3d, and 5d post-reperfusion (Figure 2). Compared to the Sham group, GSH/GSSG levels exhibited a down-regulation at the onset of reperfusion, followed by an up-regulation at three days. MDA, a pivotal end product of lipid peroxidation, has emerged as a key metric for assessing lipid peroxidation in iron-induced cell death. Notably, MDA levels in the brain tissues of mice spiked significantly at 1 day post-MCAO/R compared to the control group, subsequently demonstrating a marked recovery by the 3rd day and a return to normal levels by the 5th day. These findings underscore the peak oxidative stress incidence at the 24-hour mark post-MCAO/R. Furthermore, Fe (2+), a pivotal indicator of iron-induced cell death, exhibited substantial alterations on the first day, markedly deviating from the Sham group; by the fifth day, Fe (2+) levels no longer diverged from those of the Sham group, denoting a restoration to normal.
Changes in MCAO/R-induced ferroptosis in mice
Oxidative stress plays a crucial role in neuronal damage during CIRI. NRF2, GPX4, XCT, and HO-1 are the primary factors involved in oxidative stress and play key roles in ferroptosis. In our study, we assessed the changes in ferroptosis at the protein level on different days after MCAO/R. The results showed that, compared to the control group, the expression of NRF2, XCT, and GPX4 was significantly up-regulated on the first day and then decreased, whereas HO-1 exhibited the opposite trend (Figure 3).
Inhibiting the onset of ferroptosis enhances neuroprotection in MCAO
As shown in Figure 4A, the behavioral score of the mice showed the successful establishment of the mouse MCAO model. Furthermore, compared with the MCAO/R+ erastin group and the model group, the behavior of mice in the MCAO/R+ ferrostain-1 group showed some improvement, indicating that cerebral ischemia and reperfusion damage in mice can be mitigated by inhibiting ferroptosis. Moreover, the results of TTC (Figure 4B and C) staining showed that, compared with the sham operation group, the infarct size was significantly larger in MCAO/R mice and was dramatically reduced in the MCAO/R+ ferrostain-1 group (P<0.05). Furthermore, mouse brain tissues were collected after sacrifice and stained with hematoxylin and eosin. No obvious pathological changes were observed in the sham group. As shown in Figure 4D, at 24 hr after MCAO, neuronal pyknosis and dissolution, cell body shrinkage, swelling of neurons and glial cells in the surrounding tissues of the infarction area, and enlargement of nerve cells and perivascular space were observed, which were milder in the MCAO/R+ ferrostain-1 group, but more aggravated in the MCAO/R+ erastin group. These results indicate that inhibiting the onset of ferroptosis enhances neuroprotection in MCAO.
Discussion
Ischemic stroke has a high incidence, disability rate, mortality rate, recurrence rate, and economic burden, making it one of the major diseases threatening human health (31). Currently, the FDA has only approved intravenous tissue plasminogen activator and endovascular thrombectomy as treatment methods for IS. However, these two treatment methods must be used within 4.5 hr and 12 hr after a stroke, respectively, which is very urgent (32, 33). Reperfusion of blood after thrombus removal can also cause damage to patients, known as CIRI. However, the mechanism of injury caused by cerebral ischemia/reperfusion is complex and not fully understood, involving multiple signaling pathways (34-36). Therefore, clarifying the signaling pathways of cerebral ischemia-reperfusion injury is essential for improving CIRI.
Ferroptosis is one of the mechanisms that cause CIRI. Ferroptosis is a novel programmed cell death mode, characterized by excessive iron deposition inducing an increase in reactive oxygen species (ROS) within the cell, depletion of intracellular antioxidant glutathione (GSH), and a decrease in expression of glutathione peroxidase-4 (GPX4) that clears lipid ROS, leading to cellular toxicity (22, 37). The results of this experiment show a significant decrease in GSH and GPX4 after CIRI, indicating that ferroptosis injury occurred after CIRI. Furthermore, some studies have reported an association between over-iron death and CIRI. However, the dynamic monitoring of iron death-related pathways after CIRI has not been investigated (20, 19). Therefore, we dynamically monitored the ferroptosis-related pathways after CIRI and clearly observed a dynamic process of ferroptosis. This dynamic process further confirmed that the neuroprotection against cerebral ischemia and reperfusion injury can be strengthened by regulating the iron-related death pathway. This lays a solid theoretical foundation for the development of innovative therapeutic interventions for CIRI.
Finally, we further verify the link between ferroptosis and CIRI, TTC and HE results showed that ferroptosis inhibitors can improve the neurotoxicity of cerebral ischemia reperfusion, while ferroptosis inducer aggravated the neurotoxicity of cerebral ischemia reperfusion injury, further indicating that the neuroprotective effect of cerebral ischemia reperfusion injury can be realized by regulating the expression of ferroptosis pathway, which provides essential theoretical support to improve the survival rate of neuronal cells by inhibiting cell ferroptosis.
Middle cerebral artery occlusion and reperfusion leads to disruption of the blood brain barrier
MCAO/R leads to a decrease in GSH and Fe (2+) secretion and an up-regulation of MDA
Changes in MCAO/R-induced ferroptosis in rats
Inhibiting ferroptosis onset enhances neuroprotection in middle cerebral artery occlusion, as evidenced by reduced infarct volume and improved behavioral outcomes
Graphical Abstract
Conclusion
Overall, ferroptosis was closely related to CIRI (Figure 5). Dynamic monitoring of ferroptosis following brain ischemia-reperfusion injury revealed a specific therapeutic time window for ferroptosis-related CIRI, providing a theoretical basis for enhancing neuronal cell survival through ferroptosis inhibition.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Martins SO Mont’Alverne F Rebello LC Abud DG Silva GS Lima FO Thrombectomy for stroke in the public health care system of Brazil N Engl J Med 2020382231623263252113310.1056/NEJ Moa 2000120 · doi ↗ · pubmed ↗
- 2Thayabaranathan T Kim J Cadilhac DA Thrift AG Donnan GA Howard G Global stroke statistics Int J Stroke 2022179469563597598610.1177/17474930221123175 PMC 9980380 · doi ↗ · pubmed ↗
- 3Grysiewicz RA Thomas K Pandey DK Epidemiology of ischemic and hemorrhagic stroke: Incidence, prevalence, mortality, and risk factors Neurol Clin 2008268718951902689510.1016/j.ncl.2008.07.003 · doi ↗ · pubmed ↗
- 4Barthels D Das H Current advances in ischemic stroke research and therapies Biochim Biophys Acta Mol Basis Dis 202018661652601652863169936510.1016/j.bbadis.2018.09.012PMC 6981280 · doi ↗ · pubmed ↗
- 5Sommer CJ Ischemic stroke: Experimental models and reality Acta Neuropathol 20171332452612806435710.1007/s 00401-017-1667-0PMC 5250659 · doi ↗ · pubmed ↗
- 6Maida CD Norrito RL Daidone M Tuttolomondo A Pinto A Neuroinflammatory mechanisms in ischemic stroke: Focus on cardioembolic stroke, background, and therapeutic approaches Int J Mol Sci 202021645464873289961610.3390/ijms 21186454 PMC 7555650 · doi ↗ · pubmed ↗
- 7Liu R Li Y Zhang X Tang K Li J Fang T Applications and mechanisms of taohong siwu decoction in the treatment of ischemic stroke Pharmacogn Mag 20242010711085
- 8Yang K Zeng L Yuan X Wang S Ge A Xu H The mechanism of ferroptosis regulating oxidative stress in ischemic stroke and the regulation mechanism of natural pharmacological active components Biomed Pharmacother 20221541136111136333608128810.1016/j.biopha.2022.113611 · doi ↗ · pubmed ↗