Recurrent Optic Neuritis With MRI-Negative Myelopathy in Myelin Oligodendrocyte Glycoprotein Antibody-Associated Disease (MOGAD)
Karishma Harrilal-Maharaj, Branimir Nevajda

TL;DR
A case study shows how optic neuritis can signal a rare neurological disease even when MRI results are normal.
Contribution
Highlights the diagnostic challenge of MOGAD when MRI is negative and emphasizes early treatment to prevent disability.
Findings
A patient with MOGAD showed optic neuritis and myelopathy symptoms despite normal spinal MRI.
Early treatment with corticosteroids and plasma exchange led to partial recovery.
Recurrent optic neuritis can be an early sign of MOGAD even with normal imaging.
Abstract
Myelin oligodendrocyte glycoprotein antibody-associated disease (MOGAD) is an inflammatory demyelinating disorder that frequently presents with optic neuritis or myelitis, although early diagnostic evaluation may be challenging when neuroimaging is normal despite significant neurological deficits. We describe a 29-year-old woman who initially developed unilateral optic neuritis, followed one month later by sequential contralateral involvement and new sensory disturbances. She subsequently presented with acute bilateral lower-limb weakness, brisk reflexes, and lumbosacral sensory disturbance, although spinal MRI remained normal. Progressive neurological decline prompted treatment with high-dose IV corticosteroids and plasma exchange. MOG-IgG later returned positive, confirming MOGAD. The patient experienced gradual recovery of left-eye color perception and lower-limb strength, whereas…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
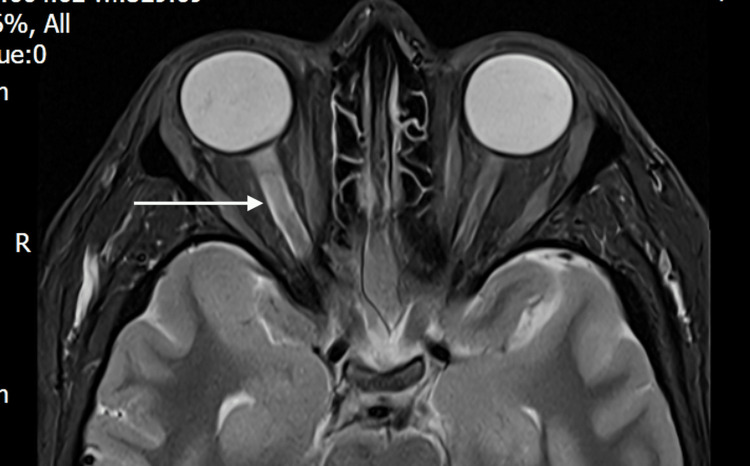 Figure 1
Figure 1| Time point | Key clinical features | Investigations | Management |
| Late August 2025 | Right-eye visual blurring; intermittent bilateral hand paresthesia | MRI brain/orbits: right optic neuritis; no intracranial lesions | No corticosteroids |
| September 2025 | Sequential left optic neuritis; painful eye movements (horizontal gaze); severe visual loss | MRI brain/orbits: persistent right optic neuritis; subtle left optic nerve involvement; no perineural enhancement | IV methylprednisolone ×3 days, oral taper |
| Early October 2025 | Bilateral lower-limb weakness; paresthesia; shock-like lumbosacral sensations; hyperreflexia | MRI brain and spine: no spinal cord lesions; CSF: elevated protein, normal cell count, OCB negative | IV methylprednisolone; tertiary referral |
| Mid-October 2025 | Progressive neurological symptoms | Serum MOG-IgG positive (1:10); AQP4-IgG negative | PLEX ×5 sessions |
| Early November 2025 | Improved left-eye color vision and lower-limb strength; persistent right-eye visual loss | - | Ongoing oral steroid taper |
| Feature | MOGAD | AQP4-NMOSD |
| Primary target | Myelin oligodendrocyte glycoprotein (oligodendrocytes) | AQP4 (astrocytes) |
| Optic neuritis pattern | Often bilateral/sequential; disc edema; better recovery | Often severe, profound, sometimes chiasmal; poorer recovery |
| Myelitis | May be MRI-negative early; good recovery in many cases | Typically longitudinally extensive; severe, with early bladder involvement |
| Steroid responsiveness |
Typically strong, rapid improvement [ |
Often incomplete; PLEX frequently required [ |
| Effect of PLEX |
Substantial visual/neurological recovery [ |
Helpful but often with residual disability [ |
| Long-term immunotherapy |
Individualized (monophasic vs. relapsing) [ |
Essential in all AQP4-positive patients [ |
| Relapse pattern |
Monophasic or relapsing; relapse risk variable [ |
Highly relapsing, often severe [ |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsMultiple Sclerosis Research Studies · Peripheral Neuropathies and Disorders · Autoimmune Neurological Disorders and Treatments
Introduction
Myelin oligodendrocyte glycoprotein antibody-associated disease (MOGAD) is an immune-mediated demyelinating disorder of the CNS characterized by optic neuritis, myelitis, and, less commonly, acute disseminated encephalomyelitis [1-3]. Although it shares overlapping features with multiple sclerosis (MS) and aquaporin-4-positive neuromyelitis optica spectrum disorder (AQP4-NMOSD), MOGAD represents a distinct pathological and clinical entity. Patients typically demonstrate more pronounced optic nerve involvement, a monophasic or relapsing course, and a characteristic response to corticosteroids, while lacking the chronic neurodegenerative features associated with MS [1-3].
Diagnosing MOGAD can be challenging in the acute setting. MRI abnormalities may be subtle or even absent despite clinically significant inflammation, particularly in cases of early myelitis [4,5]. This can lead to diagnostic uncertainty, especially when symptoms evolve rapidly or do not conform to classical MS or NMOSD patterns. Recurrent or sequential optic neuritis, bilateral involvement, steroid responsiveness, and disproportionately severe visual loss should raise suspicion for MOGAD even when neuroimaging is nondiagnostic [4,6].
We describe a young woman who developed sequential bilateral optic neuritis followed by clinically evident myelopathy in the context of a normal spinal MRI. This case highlights the diagnostic pitfalls associated with MRI-negative MOGAD and underscores the importance of a clinically driven approach, supported by timely antibody testing and early therapeutic intervention. While isolated reports of MRI-negative myelitis exist, the combination of rapidly sequential bilateral optic neuritis followed shortly thereafter by clinically significant but radiologically silent myelopathy is relatively uncommon in adults. This pattern can create substantial diagnostic uncertainty, particularly early in the disease course.
By presenting this case, we aim to draw attention to this under-recognized phenotype, illustrate the limitations of spinal MRI in early MOGAD, and reinforce the need for clinical suspicion to prompt early antibody testing and treatment escalation. The objective of this report is therefore to support clinicians in identifying similar presentations and preventing delays that may contribute to poorer visual or neurological outcomes.
Case presentation
A 29-year-old woman with a history of migraines presented to ophthalmology in late August 2025 with blurred vision in her right eye. She also reported a one-year history of intermittent paresthesia in both hands. On initial ophthalmic assessment, the right optic disc appeared normal, with no disc edema (grade 0), consistent with retrobulbar optic neuritis. Color vision testing using Ishihara plates was mildly reduced in the right eye (22/24) and normal in the left (24/24), with no relative afferent pupillary defect at this stage. Extraocular movements were full, with pain reported on eye movement in multiple directions, most prominent on horizontal gaze.
MRI of the brain and orbits performed one week later demonstrated high signal within the right optic nerve, consistent with optic neuritis (Figure 1), with no additional abnormalities. There was no evidence of optic nerve sheath or perineural enhancement, no optic chiasmal involvement, and no intracranial parenchymal lesions. She was discharged without corticosteroid therapy.
Axial T2-weighted fat-suppressed MRI of the orbits, performed shortly after symptom onset, showing hyperintensity and swelling of the right optic nerve (arrow) consistent with acute optic neuritis, with no additional intracranial abnormalities
She re-presented to the ED one month after her initial presentation with sudden visual loss in the left eye, accompanied by painful eye movements and a severe headache resembling her typical migraine. Eye pain preceded the visual loss and was again most noticeable on horizontal gaze. Examination revealed no perception of light in the right eye and a visual acuity of 6/24 in the left eye. A right relative afferent pupillary defect was now present, and color vision was markedly reduced in the right eye. She also described tingling in the right upper limb and intermittent facial paresthesia. She was initially reviewed by the stroke team, with no focal cortical or brainstem signs identified, and was subsequently assessed in the eye casualty, where left-eye visual acuity, color vision, pupillary responses, and fundoscopic findings were unchanged from the earlier assessment. Owing to her progressive symptoms, she was admitted for medical evaluation. A repeat MRI of the brain and orbits demonstrated persistent right optic nerve swelling and a subtle high signal in the posterior segment of the left optic nerve; the brain parenchyma remained normal. No perineural or optic nerve sheath enhancement was identified. She received three days of IV methylprednisolone followed by a five-day oral taper.
In early October, approximately six weeks after her initial presentation, she attended another ED within the same trust, reporting new bilateral lower-limb weakness, intermittent tingling, and “shock-like” sensations in both legs, more prominent in a lumbosacral distribution. She also noted transient numbness of the posterior left thigh. Examination revealed mild bilateral lower-limb weakness and brisk reflexes, with preserved coordination and no sensory level.
An MRI of the brain and entire spine performed during this admission showed interval improvement of the right optic neuritis but no signal abnormality within the spinal cord. Lumbar puncture demonstrated elevated protein (0.77 g/L) with normal cell counts and negative oligoclonal bands; viral and bacterial studies were also negative. This CSF profile, characterized by elevated protein with normal cell counts and negative oligoclonal bands, in the context of sequential optic neuritis and clinically evident but MRI-negative myelopathy, was felt to be more supportive of MOGAD than MS or AQP4-positive NMOSD.
Due to ongoing neurological deterioration, neurology recommended a further course of IV methylprednisolone and arranged a consultation with a tertiary neuroscience center, which accepted her for plasma exchange (PLEX). She underwent five sessions of PLEX between October 13 and 17, 2025, and was discharged on a tapering course of oral prednisolone.
Serum MOG-IgG and AQP4-IgG were sampled during the tertiary admission, after corticosteroid treatment, and prior to initiation of PLEX. AQP4-IgG was negative, while MOG-IgG returned positive at a titer of 1:10 using a cell-based assay, confirming the diagnosis of MOGAD. At follow-up in early November, she reported gradual improvement in left-eye color perception and lower-limb strength, although right-eye visual acuity remained profoundly reduced. She also described a further episode of back pain and transient weakness despite ongoing steroid therapy.
A chronological summary of the patient’s clinical course, investigations, and management is provided in Table 1.
Discussion
MOGAD encompasses a spectrum of inflammatory demyelinating disorders characterized by optic neuritis, myelitis, or acute disseminated encephalomyelitis [1-3]. Unlike MS or AQP4-NMOSD, MOGAD often demonstrates steroid responsiveness, less chronic neurodegeneration, and distinct radiological patterns [1-3]. Optic neuritis represents the most common presentation, occurring in up to 60-80% of cases, frequently with bilateral or sequential involvement and often associated with substantial optic disc edema [4-6]. Myelitis is similarly well recognized, but MRI-negative myelopathy remains an underappreciated diagnostic pitfall [4,5].
MRI-negative myelopathy in MOGAD
Although spinal cord involvement typically produces T2-hyperintense lesions on MRI, up to 10-15% of patients with MOGAD myelitis may demonstrate normal early spinal imaging despite clear clinical signs of cord dysfunction [4,5]. Proposed explanations include transient inflammatory changes below the threshold of radiological detection, reversible conduction block, or very early disease presentation before radiological evolution. These cases highlight the limitations of relying solely on MRI for diagnosis in demyelinating conditions.
Our patient exhibited bilateral lower-limb weakness, hyperreflexia, and lumbosacral sensory disturbance, findings strongly suggestive of myelopathy, yet her spinal MRI remained normal. The coexistence of recent optic neuritis further strengthened the suspicion of an inflammatory demyelinating process. This case reinforces that a normal spinal MRI does not exclude clinically significant myelopathy in MOGAD, and clinicians should maintain a high index of suspicion when neurological findings and recent optic neuritis coexist.
Diagnostic complexity and evolving clinical course
MOGAD frequently evolves in a stepwise pattern, with individual relapses affecting different regions of the CNS [1,3,6]. In this case, the patient’s clinical course progressed from unilateral optic neuritis to sequential bilateral involvement, then to acute myelopathic symptoms within a six-week period. Early symptoms lacked widespread neurological involvement, and brain MRI remained normal, which can obscure the broader diagnosis [4,5]. Indeed, early presentations may mimic MS, idiopathic optic neuritis, or even migraine-associated visual disturbance, particularly in young adults [4-6]. CSF findings may assist in differentiation, as MOGAD often shows elevated protein with negative oligoclonal bands, unlike MS [7,8]. In this case, MOG-IgG was detected using a cell-based assay at a low positive titer (1:10), which was interpreted as clinically meaningful in the context of recurrent optic neuritis, MRI-negative myelopathy, and recent corticosteroid exposure, which is recognized to reduce circulating antibody titers.
Studies have shown that sequential or bilateral optic neuritis, severe visual loss, and pain with eye movement should prompt early consideration of MOGAD, even in the absence of definitive MRI findings [6]. Early neurology involvement is crucial, particularly when symptoms evolve rapidly or when atypical features such as bilateral optic neuritis or steroid-responsive relapses are present. This case demonstrates precisely how the clinical picture, rather than imaging alone, should guide investigation and acute management.
Treatment implications
High-dose corticosteroids remain the first-line treatment for acute MOGAD attacks, with many patients showing rapid improvement [9,10]. However, up to one-third of patients may experience steroid-refractory disease requiring escalation to PLEX or IV immunoglobulin [11]. The decision by the tertiary center to initiate PLEX before the MOG-IgG result returned aligns with recommendations for severe or progressive optic neuritis and myelitis, particularly when visual acuity is significantly impaired [11,12].
Our patient experienced partial neurological recovery following PLEX, consistent with reported outcomes in the literature [12], but persistent right-eye visual loss underscores that delayed recognition or incomplete early treatment may be associated with poorer visual prognosis [9,10]. Long-term relapse rates in MOGAD may reach 30-50%, and recurrent attacks can lead to cumulative disability, highlighting the need for ongoing specialist follow-up and consideration of maintenance immunotherapy in selected patients [13].
Comparison of MOGAD and AQP4-positive NMOSD
Although MOGAD and AQP4-positive NMOSD can present with overlapping features such as optic neuritis and longitudinally extensive myelitis, they represent distinct autoimmune disorders with important diagnostic and therapeutic implications [2,3]. NMOSD is driven by AQP4-IgG antibodies targeting astrocytes, whereas MOGAD reflects an oligodendrocytopathy with immune targeting of myelin oligodendrocyte glycoprotein [2,3]. Optic neuritis in NMOSD often causes profound and irreversible visual impairment with frequent chiasmal or optic tract involvement, while MOGAD more commonly presents with bilateral or sequential optic neuritis, marked optic disc edema, and generally better visual recovery following corticosteroid treatment [4-6,9,10]. Myelitis in NMOSD is typically severe, longitudinally extensive, and associated with early sphincter dysfunction and high relapse risk, whereas MOGAD myelitis may demonstrate better functional outcomes and can be MRI-negative in the early disease course [4,5]. Treatment responses also differ significantly: MOGAD generally shows a strong response to corticosteroids, and PLEX often yields substantial recovery in severe attacks, while NMOSD relapses frequently leave residual deficits despite aggressive therapy [9-12]. Long-term immunosuppression is essential for all AQP4-positive NMOSD patients due to its highly relapsing nature, whereas maintenance therapy in MOGAD is individualized based on relapse frequency, attack severity, and functional outcomes [11-13]. A comparison of the key clinical and treatment differences between MOGAD and AQP4-positive NMOSD is summarized in Table 2.
Broader clinical and system-level considerations
This case also illustrates the importance of robust clinical pathways for inflammatory optic neuropathies. Clear guidance on when to involve neurology, when to initiate extended antibody testing, and when to escalate treatment may reduce delays in diagnosis. Given that MRI-negative myelopathy and evolving optic neuritis can lead to diagnostic uncertainty, reliance on multidisciplinary decision-making rather than radiological findings alone is essential.
Patient perspective
The patient expressed significant frustration at the continued, low-grade progression of her symptoms, particularly the persistent visual disturbance in her right eye. She described how this impairment limits her ability to carry out day-to-day activities, especially tasks requiring prolonged computer use, resulting in frequent breaks and reduced confidence at work. She also reported ongoing anxiety about the risk of relapse and the possibility of losing vision in her left eye, which has been a major source of psychological distress.
She felt that certain aspects of her early care could have been improved. In her view, a more detailed assessment of her symptoms, particularly differentiating new neurological features from her usual migraine-related complaints, might have prompted earlier treatment. She also felt that clearer explanations regarding the significance of optic neuritis and the potential differential diagnoses, including conditions such as NMOSD or MOGAD, would have enabled her to advocate more strongly for urgent therapy at the time.
Looking back, she recognized that several early symptoms, such as stiffness and weakness in her legs and back, intermittent paresthesia, fatigue, and fluctuating visual blurriness, were more significant than she had initially appreciated, but she had attributed them to migraines or lifestyle factors. She reflected that greater awareness of the potential implications of optic neuritis may have influenced how she interpreted and escalated these symptoms.
Her reflections highlight the emotional, functional, and psychological impact of MOGAD and underscore the importance of early recognition, clear communication, and patient-centered care, not only in inflammatory neuropathies but across all medical conditions.
Conclusions
This case illustrates the diagnostic challenges posed by MOGAD, particularly when evolving neurological symptoms occur in the context of normal spinal imaging. Recurrent or bilateral optic neuritis should alert clinicians to the possibility of MOGAD, even when MRI findings appear limited or nondiagnostic. The development of clinically evident myelopathy despite a normal spinal MRI underscores the need to prioritize the clinical examination over radiological appearances in early disease. Timely MOG-IgG testing and early escalation to treatments such as high-dose corticosteroids and PLEX are essential to prevent irreversible visual and neurological morbidity. Ultimately, this case reinforces the importance of recognizing atypical inflammatory demyelinating presentations and adopting a clinically driven approach to diagnosis and management.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Myelin oligodendrocyte glycoprotein antibodies in neurological disease Nat Rev Neurol Reindl M Waters P 89102152019 https://pubmed.ncbi.nlm.nih.gov/30559466/3055946610.1038/s 41582-018-0112-x · doi ↗ · pubmed ↗
- 2Clinical features and risk of relapse in children and adults with myelin oligodendrocyte glycoprotein antibody-associated disease Ann Neurol Cobo-Calvo A Ruiz A Rollot F 30418920213295942710.1002/ana.25909 · doi ↗ · pubmed ↗
- 3MOG encephalomyelitis: international recommendations on diagnosis and antibody testing J Neuroinflammation Jarius S Paul F Aktas O 134152018 https://jneuroinflammation.biomedcentral.com/articles/10.1186/s 12974-018-1144-22972422410.1186/s 12974-018-1144-2PMC 5932838 · doi ↗ · pubmed ↗
- 4Clinical presentation and prognosis in MOG-antibody disease: a UK study Brain Jurynczyk M Messina S Woodhall MR 312831381402017 https://doi.org/10.1093/brain/awx 2762913609110.1093/brain/awx 276 · doi ↗ · pubmed ↗
- 5Radiological characteristics of myelin oligodendrocyte glycoprotein antibody disease Mult Scler Relat Disord Salama S Khan M Levy M Izbudak I 1522292019 https://doi.org/10.1016/j.msard.2019.01.0213065825910.1016/j.msard.2019.01.021PMC 6431795 · doi ↗ · pubmed ↗
- 6Clinical phenotype, radiological features, and treatment of myelin oligodendrocyte glycoprotein-immunoglobulin G (MOG-Ig G) optic neuritis Curr Opin Neurol Chen JJ Bhatti MT 4754332020 https://journals.lww.com/co-neurology/abstract/2020/02000/clinical_phenotype,_radiological_features,_and.9.aspx 3174323510.1097/WCO.0000000000000766 · doi ↗ · pubmed ↗
- 7MOG-Ig G in NMO and related disorders: a multicenter study of 50 patients. Part 2: epidemiology, clinical presentation, radiological and laboratory features, treatment responses, and long-term outcome J Neuroinflammation Jarius S Ruprecht K Kleiter I 280132016 https://link.springer.com/article/10.1186/s 12974-016-0718-02779320610.1186/s 12974-016-0718-0PMC 5086042 · doi ↗ · pubmed ↗
- 8Autoantibodies to MOG in a distinct subgroup of adult multiple sclerosis Neurol Neuroimmunol Neuroinflamm Spadaro M Gerdes LA Krumbholz M 032016 https://doi.org/10.1212/NXI.000000000000025710.1212/NXI.0000000000000257 PMC 494977527458601 · doi ↗ · pubmed ↗