Hemophagocytic Lymphohistiocytosis as First Manifestation of Dual B‐Cell Neoplasms: A Case Report of Co‐Existing Multiple Myeloma and B‐Cell Lymphoma
Carla Romagnoli, Alexandra Lyubimova, Leily Santos‐Carrion, Ferial Alloush, Vathany Sriganeshan, Andrea Noboa, Jacqueline C. Barrientos

TL;DR
A rare case of immune disorder HLH was caused by two blood cancers in a patient, and treatment with Tocilizumab was effective.
Contribution
This is the first reported case of HLH associated with co-existing multiple myeloma and B-cell lymphoma.
Findings
HLH was driven by two distinct malignant clones in the patient's bone marrow.
Tocilizumab led to an excellent clinical and laboratory response.
Initial therapies like steroids and chemotherapy failed to produce results.
Abstract
Hemophagocytic lymphohistiocytosis (HLH) is an immune disorder causing excessive inflammation and tissue damage. Lymphoma, especially T‐cell lymphoma, is the most common cause of HLH, while Multiple Myeloma (MM) is rarely associated. We present a 61‐year‐old man with spiking fevers, fatigue, and unintentional weight loss. The HLH was driven by two distinct malignant clones in his bone marrow: plasma cell neoplasm and B‐cell lymphoma. Patient failed initial therapy with steroids, anakinra, and chemotherapy. Tocilizumab use, on the other hand, led to an excellent clinical and laboratory response immediately. This is the first reported case of HLH associated with two hematological malignancies. Trial Registration: The authors have confirmed clinical trial registration is not needed for this submission
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
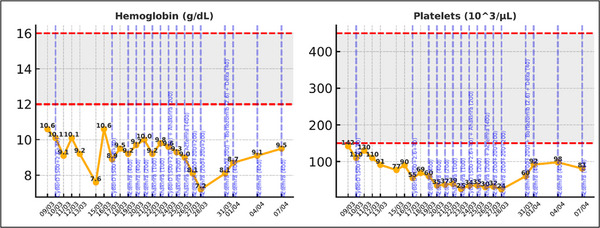 FIGURE 1
FIGURE 1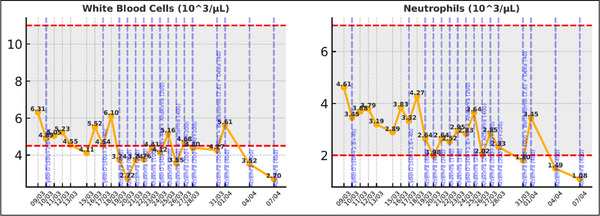 FIGURE 2
FIGURE 2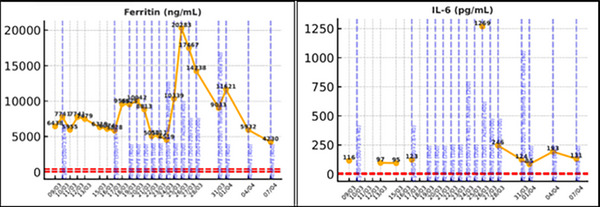 FIGURE 3
FIGURE 3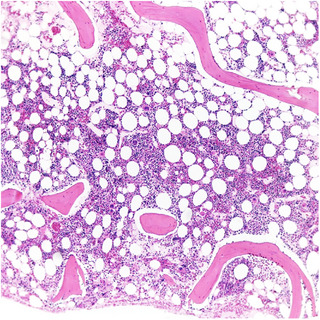 FIGURE 4
FIGURE 4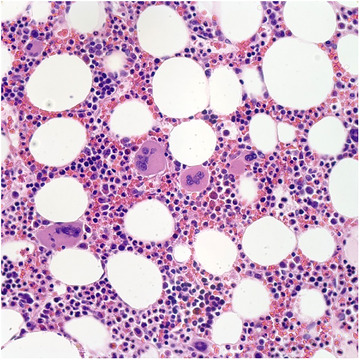 FIGURE 5
FIGURE 5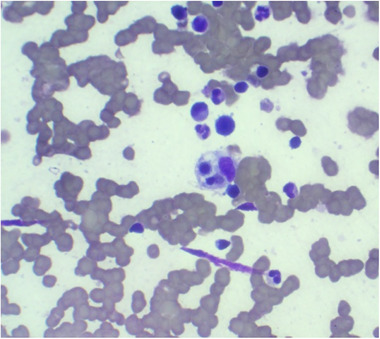 FIGURE 6
FIGURE 6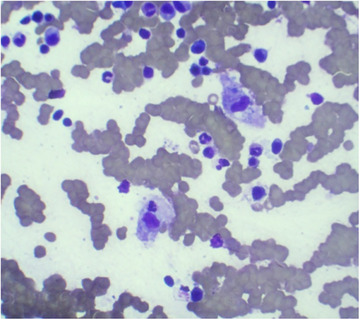 FIGURE 7
FIGURE 7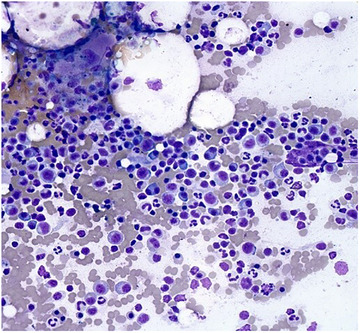 FIGURE 8
FIGURE 8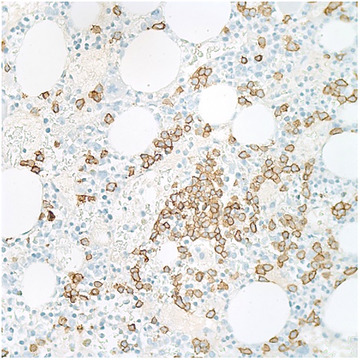 FIGURE 9
FIGURE 9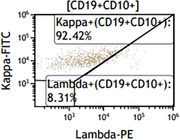 FIGURE 10
FIGURE 10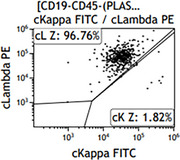 FIGURE 11
FIGURE 11| Age (in years)/sex | Type of MM | HLH criteria | Possible triggers | HLH‐directed therapy | Chemotherapy | Outcome | Reference |
|---|---|---|---|---|---|---|---|
| 59/M | IgA Lambda |
Fever Hemophagocytosis Hyperferritinemia Increased sCD25 Hypertriglyceridemia | POD | no | DVR‐PACE | Bad response, switch to DP. Died after 5 months from POD | [ |
| 56/M | IgG Lambda |
Fever Bicytopenia Hyperferritinemia Hemophagocytosis Increased sCD25 | Not given | Dexamethasone and etoposide | No | Multiorgan failure after 8 weeks of treatment | [ |
| 29/not given | IgG Kappa |
Fever Pancytopenia Hypofibrinogenemia Hyperferritinemia Hemophagocytosis |
Lenalidomide related MM progression | Dexamethasone and etoposide |
Withhold lenalidomide KD‐PACE Allo‐HSCT | HLH resolved shortly with quick POD | [ |
| 64/M | IgA Kappa |
Fever Pancytopenia Splenomegaly Hyperferritinemia Hemophagocytosis | Disease | Methylprednisolone | Cyclophosphamide and dexamethasone | Multiorgan failure. Died after 15 days of hospitalization | [ |
| 59/M | IgG Lambda |
Fever Pancytopenia Hyperferritinemia Increased sCD25 |
Respiratory syncytial virus Incomplete immune reconstitution after ASCT | Dexamethasone, tocilizumab and anakinra | Carfilzomib and daratumumab | On Day 4 of anakinra patient developed septic shock and passed away | [ |
| 78/M | IgA Kappa |
Fever Bicytopenia Hyperferritinemia Hypofibrinogenemia Hemophagocytosis Low NK cell activity Increased sCD25 |
EBV infection. disease | Dexamethasone and anakinra | Cyclophosphamide, bortezomib and dexamethasone | Six months later VGPR. Normalized blood counts | [ |
| Age | 61 |
| Gender | Male |
| Ethnicity | Hispanic/Latino |
| Symptoms experienced | Fever, unintentional weight loss, fatigue |
| HIV status | Negative |
| EBV (PCR) | Detected |
| Hgb | 7.2 (14–18) |
| Neutrophils count | 3.3 (1.8–7.2) |
| Platelet count | 2400 (150–450) |
| Calcium | 8.9 (8.5–10.1) |
| Triglycerides | 405 mg/dL (30–150) |
| IL‐6 levels | 116 pg/mL (< 5.00) |
| CD25s | 21,875 pg/mL (532–1,891) |
| Ferritin | 14,238 ng/mL (26–388) |
| Beta‐2‐microglobulin | 4.010 mg/dL (1.09–2.53) |
| LDH | 1,375 U/L (81–246) |
| Creatinine | 1.10 mg/dl (0.7–1.3) |
| Kappa/Lambda ratio | 0.17 (0.26–1.65) |
| Immunofixation | IgG lambda monoclonal protein, 0.96 g/dL |
| PET‐CT scan | No hypermetabolic adenopathy or bone lesions. Splenomegaly 13.9 cm |
|
1st Biopsy (February 14, 2025) |
2nd Biopsy (March 5, 2025) | |
|---|---|---|
| Pathology | Hypercellular bone marrow for age with trilineage hematopoiesis and megakaryocytic hyperplasia (Figures | Hemophagocytosis, along with a hypercellular bone marrow demonstrating 20%–30% of plasma cells (Figures |
| Flow cytometry | − |
Two clones: ‐IgG Lambda monoclonal plasma cells ‐small subset of CD10+ kappa‐restricted monoclonal B cells. (Figures |
| Karyotype | − | 46 XY (20) |
| FISH analysis | − |
Presence of t(11;14) (Plasma cell enrichment was performed using CD138 coated beads) NEGATIVE for 13q deletion, 1p32/1q21, p53, t(4;14), t(14;16), and t(14;20). |
| Molecular assay | − | MYD88 negative |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsAutoimmune and Inflammatory Disorders Research · Otitis Media and Relapsing Polychondritis · Immunodeficiency and Autoimmune Disorders
Introduction
1
Hemophagocytic lymphohistiocytosis (HLH) is an immune response syndrome marked by aberrant hyperinflammation, hyperferritinemia, and a potentially fatal cytokine storm, marked by persistent activation of CD8+ cytotoxic T lymphocytes and natural killer (NK) cells, leading to increased secretion of inflammatory cytokines and macrophage activation resulting in excessive inflammation and tissue destruction [1, 2].
It can be divided into two main forms: primary or hereditary form (usually a pediatric disease associated with autosomal recessive inheritance, caused by loss‐of‐function mutations in genes involved in the cytotoxic function of NK cells and CD8+T cells) and secondary forms (commonly associated with conditions with immune dysregulation, such as infection, malignancy, autoimmune disorders, or drug‐induced).
Malignancy‐associated HLH may occur as a result of malignancy itself or the therapeutic intervention. Lymphoma is the most common neoplastic cause, with T‐cell lymphomas being more frequent than B‐cell lymphomas, whereas only 0.24% of cases are associated with multiple myeloma (MM) [3, 4, 5]. HLH diagnosis requires five out of nine diagnostic criteria (“HScore”): fever; splenomegaly; cytopenias affecting two or three lineages (hemoglobin < 9 g/dL, platelets < 100,000/µL, and neutrophils < 1000/µL); hypertriglyceridemia and/or hypofibrinogenemia; hemophagocytosis in the bone marrow, spleen, lymph nodes, or liver; hyperferritinemia; low or absent NK cell activity; and increased soluble CD25 (IL‐2r) concentration (> 2400 U/mL) [3, 6].
HLH is extremely rare (reported at 1 per 800,000 people and 1–10 per 1,000,000 children). The mortality of secondary HLH is high, but statistics are limited by the small number of reported cases (Table 1). Case series of adults treated with a variety of regimens report a 30‐day mortality of 20%–44% despite early detection and appropriate therapy. Malignancy‐associated HLH portends the worst prognosis among all HLH subtypes with a five‐year overall survival ranging from 10%–30% [7].
Case Report
2
A 61‐year‐old Ecuadorean man began experiencing fever and chills requiring hospitalization. Imaging of the chest, abdomen, and pelvis was unremarkable. A bronchoalveolar lavage demonstrated gram‐positive cocci and was treated with antibiotics, but he continued to spike fevers. Over the next 2 months, he lost 25 pounds and became fatigued. Infective endocarditis was ruled out, as well as COVID‐19 and dengue infections. His cell lines showed bicytopenia requiring blood transfusions. Because he had hypertriglyceridemia and hyperferritinemia, HLH was suspected, and he was started on high‐dose steroids while extensive workup was being done.
Clinical Findings
2.1
Initial laboratory examination at our center revealed anemia and thrombocytopenia in the setting of hyperferritinemia, hypertriglyceridemia, normal calcium, and elevated IL‐6 and IL‐2 soluble (see Table 2). Initial bone marrow biopsy performed in Ecuador showed trilineage hematopoiesis and megakaryocytic hyperplasia. We repeated a biopsy locally, which showed hemophagocytosis in the setting of myeloma (20%–30% of plasma cells by immunohistochemistry) and B‐cell lymphoma (Table 3 and Figures 4, 5, 6, 7, 8, 9, 10, 11). Along with clonal plasma cells in the bone marrow, the patient also exhibited anemia, meeting one of the CRAB criteria for MM.
Since the patient was unresponsive to high‐dose corticosteroids with poor symptom control, and met all nine of the nine diagnostic criteria included in the HScore, yielding a high pre‐test probability (96%–98%) for HLH, we recommended starting the patient on a regimen that would have activity on both myeloma and lymphoma clones: cyclophosphamide, bortezomib, and dexamethasone (CyBorD) and based on UK guidelines, recommended to start anakinra [11]. Despite these interventions, the inflammatory markers continued to rise with rapidly progressive clinical deterioration. Tocilizumab (Actemra) was initiated as salvage therapy with a response seen within 24 h (Figures 1, 2, 3). The patient was discharged home 10 days later, with an ongoing decrease in the inflammatory markers and improvement in the clinical symptoms (Supporting Information).
Evolution of Hgb and platelets following initiation of treatment.
Evolution of white blood cells following initiation of treatment.
Evolution of Ferritin and IL‐6 following initiation of treatment.
(Hematoxylin and Eosin, 5×). Hypercellular bone marrow for age with trilineage hematopoiesis.
(Hematoxylin and Eosin, 40×). Bone marrow biopsy showing increased megakaryocytes.
(Giemsa, 60×). Bone marrow aspirate smear showing hemophagocytosis (arrow).
(Giemsa, 60×). Bone marrow aspirate smear showing hemophagocytosis of a neutrophilic precursor cell (arrow).
(Giemsa, 40×). Bone marrow aspirate smear showing increased plasma cells including binucleated forms.
(CD138 IHC, 40×). Immunohistochemical stain for CD138 is positive highlighting focal increase in plasma cells, comprising 20%–30% of the marrow cellularity.
Flow cytometric analysis of the bone marrow aspirate showing a kappa‐restricted, CD10 positive monoclonal B‐cell population.
A lambda‐restricted, monoclonal plasma cell population was also identified with loss of CD19 expression.
Discussion
3
HLH associated with MM is a rare occurrence, particularly when it also involves an additional B‐cell malignancy. To our knowledge, this is the first report in the literature documenting HLH associated with two distinct untreated hematological malignancies.
One of the major challenges in treating patients with HLH is achieving a timely diagnosis. It is crucial to identify and address the underlying triggers of HLH. Despite increasing awareness among physicians and the rising incidence of HLH—especially with the growing use of immune‐activating and modulating agents—significant uncertainty still exists regarding the optimal diagnostic approach for this condition [4]. The patient met all the diagnostic criteria, which is uncommon. However, some results became available only after the therapy had been initiated, as time is of the essence when this diagnosis is high on the differential.
Various therapeutic strategies have been employed without clear management for someone with two different malignancies. Treatment typically involves a combination of HLH‐directed therapy and malignancy‐specific treatment [7]. The main goal of induction therapy is to suppress the inflammatory process. To achieve this, he was started on steroids and then followed up with chemotherapy, choosing a regimen that is effective for both myeloma and lymphoma. Anakinra, an IL‐1 antagonist that has been increasingly used in the treatment of secondary HLH, was also added [12]. Given the lack of initial response, and in coordination with the local team in Ecuador, we agreed to use tocilizumab while continuing chemotherapy. While emapalumab, an interferon‐gamma (IFN‐γ) blocking antibody, has been reported to have activity in HLH, this drug was not available in Ecuador. Tocilizumab is an anti‐IL‐6 receptor monoclonal antibody used for the management of cytokine release syndrome by inhibiting cytokines and signaling molecules involved in the inflammatory pathway [13]. While some prior reports cautioned against tocilizumab use due to limited efficacy [14, 15], treatment with tocilizumab resulted in a rapid improvement in symptomatology. Patient has started to wean off the drug and is on Cycle 2 of CyBorD.
This report highlights the rarity and complexity involved in diagnosing secondary HLH and its management. The case is significant not only for the rare diagnosis but also for the successful application of targeted therapy with tocilizumab to treat secondary malignancy‐associated HLH. This information may be of benefit to colleagues dealing with similar cases. We also would like to highlight the critical role that telemedicine played in the collaborative efforts surrounding the diagnosis and treatment of this patient, who was hospitalized abroad. Through secure and timely virtual consultations with the patient and local physician, telemedicine enabled minimizing geographical boundaries, by utilizing our collective expertise and developing an optimal care plan. This coordinated effort was instrumental in achieving the best possible outcome for the patient.
Author Contributions
A.N., J.B., and L.S.C. were involved in patient care and collected clinical information. F.A. and V.S. did pathological analysis. C.R., A.L., and J.B. wrote the manuscript. All authors critically reviewed and approved the final manuscript.
Funding
The authors have nothing to report.
Ethics Statement
The authors have nothing to report.
Consent
Consent was obtained from the patient for publication of this case report and any accompanying image.
Conflicts of Interest
Jacqueline Barrientos has received speaker honoraria from Janssen, BeiGene, and AstraZeneca; is a consultant for BeiGene, AstraZeneca, Pharmacyclics, and Janssen; and has received research support from Merk, Nurix, and Abbvie. The other authors declare no conflicts of interest.
Supporting information
Supporting Information
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1M. Al‐Ammari , D. Hsu , and A. Bryant , “Myeloma‐Associated Hemophagocytic Lymphohistiocytosis—A Comprehensive Case Study and a Novel Chemotherapy‐Free Approach With Anakinra,” EJ Haem 5, no. 5 (2024): 1057–1062.39415906 10.1002/jha 2.975PMC 11474360 · doi ↗ · pubmed ↗
- 2F. R. Mendes , K. M. Sobral , H. F. Culler , et al., “Acquired Hemophagocytic Lymphohistiocytosis as Initial Manifestation of Multiple Myeloma: A Case Report and Literature Review,” Medicine 99, no. 39 (2020): e 22299.32991435 10.1097/MD.0000000000022299 PMC 7523830 · doi ↗ · pubmed ↗
- 3E. J. Vick , K. Patel , P. Prouet , and M. G. Martin , “Proliferation Through Activation: Hemophagocytic Lymphohistiocytosis in Hematologic Malignancy,” Blood Advances 1 (2017): 779–791.29296722 10.1182/bloodadvances.2017005561 PMC 5728056 · doi ↗ · pubmed ↗
- 4N. Daver , K. Mc Clain , C. E. Allen , et al., “A Consensus Review on Malignancy‐Associated Hemophagocytic Lymphohistiocytosis in Adults,” Cancer 123 (2017): 3229–3240.28621800 10.1002/cncr.30826 PMC 5568927 · doi ↗ · pubmed ↗
- 5J. Knauft , T. Schenk , T. Ernst , et al., “Lymphoma‐Associated Hemophagocytic Lymphohistiocytosis (LA‐HLH): A Scoping Review Unveils Clinical and Diagnostic Patterns of a Lymphoma Subgroup With Poor Prognosis,” Leukemia 38 (2024): 235–249.38238443 10.1038/s 41375-024-02135-8PMC 10844097 · doi ↗ · pubmed ↗
- 6C. M. Hsu , J. M. Bennett , and B. Lipe , “Hemophagocytic Lymphohistiocytosis in a Patient With Multiple Myeloma,” Clinical Lymphoma Myeloma and Leukemia 19, no. 1 (2019): e 29–e 32.10.1016/j.clml.2018.08.01730309798 · doi ↗ · pubmed ↗
- 7A. Zoref‐Lorenz , T. E. Witzig , J. R. Cerhan , and M. B. Jordan , “Malignancy‐Associated HLH: Mechanisms, Diagnosis, and Treatment of a Severe Hyperinflammatory Syndrome,” Leukemia and Lymphoma 66, no. 4 (2024): 628–636.39656557 10.1080/10428194.2024.2436037 · doi ↗ · pubmed ↗
- 8R. Bhatt , S. Xiao , P. Gohari , and A. Podrumar , “Acquired Haemophagocytic Lymphohistiocytosis Secondary to Multiple Myeloma,” BMJ 12, no. 9 (2019): e 231084.10.1136/bcr-2019-231084 PMC 673190031488450 · doi ↗ · pubmed ↗