Characterization of the role of putative Aeromonas caviae-specific virulence factor, flgB, in virulence and host–pathogen interactions
Bernadette A. Hritzo, Jane M. Michalski, David A. Rasko

TL;DR
This study identifies and characterizes flgB, a potential virulence factor in Aeromonas caviae, showing its role in motility, adherence, and inflammation during infection.
Contribution
The study identifies flgB as a putative A. caviae-specific virulence factor and demonstrates its functional role in virulence and host interactions.
Findings
Deletion of flgB abolished swimming motility and polar flagella assembly in A. caviae.
flgB deletion reduced bacterial adherence to HT-29 cells and decreased proinflammatory cytokine production.
flgB deletion modestly attenuated virulence in a Galleria mellonella in vivo model.
Abstract
Aeromonas caviae, Gram-negative bacteria ubiquitous in the environment, are an emerging human pathogen associated with various infectious diseases, particularly gastroenteritis. Despite recent studies demonstrating A. caviae is the most predominant Aeromonas species underlying human infection, A. caviae remains understudied, and no A. caviae-specific virulence factors associated with human disease have been identified. To identify A. caviae-specific putative virulence factors, we conducted comparative genomic analyses among clinical Aeromonas isolates (n = 431), which identified a variant of flgB, predicted to encode a polar flagellum machinery protein, as over-represented in A. caviae isolates. To examine the role of flgB in virulence and host–pathogen interactions, we generated an A. caviae flgB deletion mutant and genetic complementation constructs. Swimming motility and polar…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
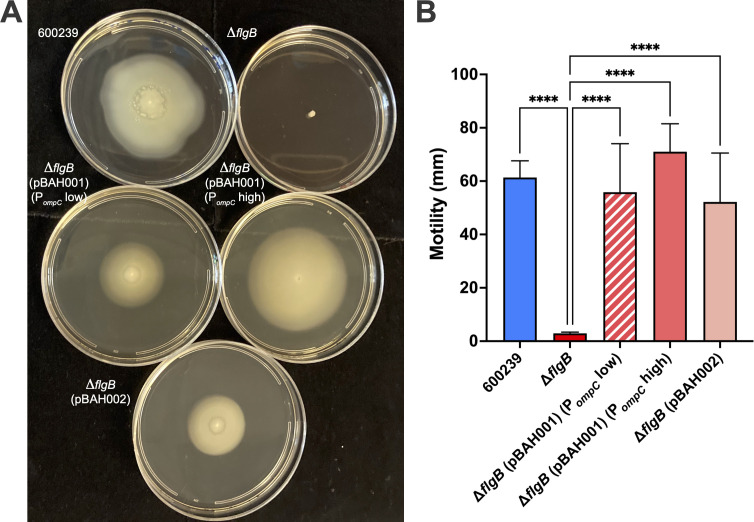 Fig 1
Fig 1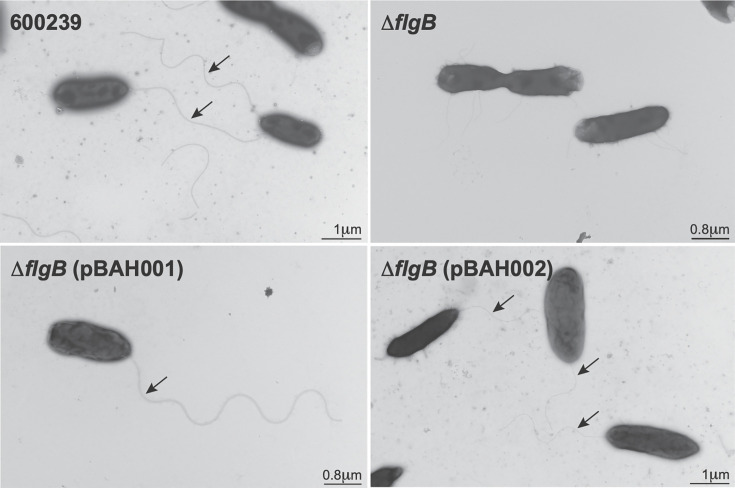 Fig 2
Fig 2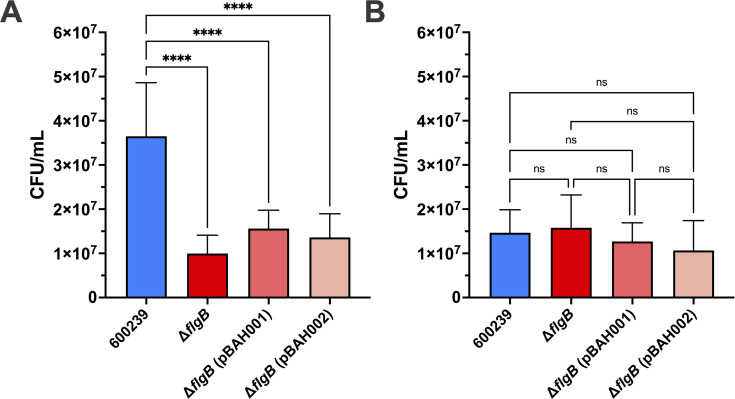 Fig 3
Fig 3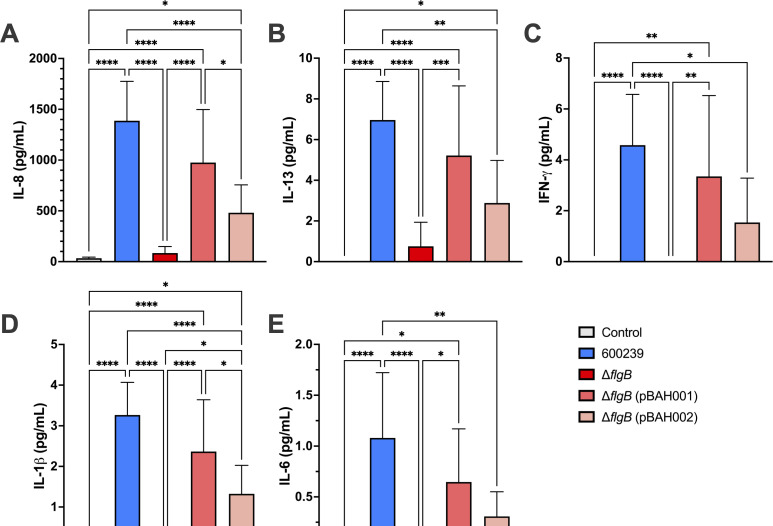 Fig 4
Fig 4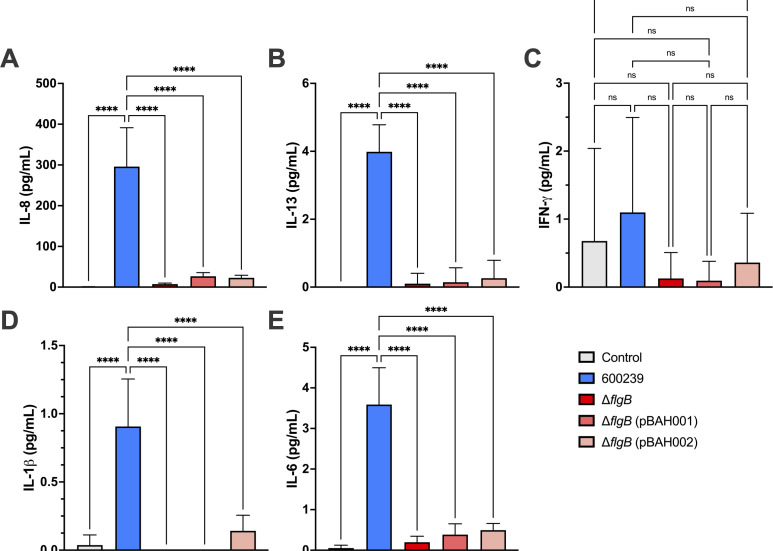 Fig 5
Fig 5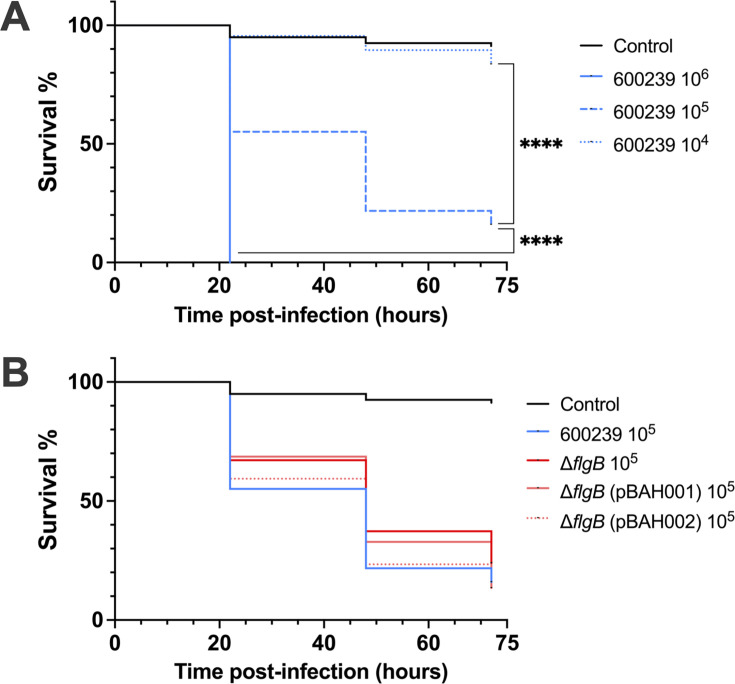 Fig 6
Fig 6| ID | Description | Reference/source |
|---|---|---|
| Strains | ||
| 600239 | Wild-type | ( |
| Δ | Stable chromosomal deletion of | This study |
| Δ | Δ | This study |
| Δ | Δ | This study |
| Plasmids | ||
| pKM200 | Lambda Red recombinase (CmR, 20 µg/mL); temperature-sensitive origin of replication (30°C) | ( |
| pKD4 | Helper plasmid containing Kanamycin/Neomycin cassette | ( |
| pCP20 | Plasmid containing flippase (CmR, 20 µg/mL); temperature-sensitive origin of replication (30°C) | ( |
| pCR-Blunt II-TOPO:: | Invitrogen pCR-Blunt II-TOPO::600239 | This study |
| pCR-Blunt II-TOPO:: | Invitrogen pCR-Blunt II-TOPO::600239 | This study |
| pSEC10 | Low copy number vector (5 copies/cell) for complementation construct; contains osmoregulated | ( |
| pSEC10-M | Low copy number vector (five copies/cell) for complementation construct; lacks osmoregulated | ( |
| pBAH001 | pSEC10::600239 | This study |
| pBAH002 | pSEC10-M::600239 | This study |
- —National Institutes of Healthhttp://dx.doi.org/10.13039/100000002
- —National Institutes of Healthhttp://dx.doi.org/10.13039/100000002
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsAquaculture disease management and microbiota · Salmonella and Campylobacter epidemiology · Vibrio bacteria research studies
INTRODUCTION
Aeromonas spp., Gram-negative bacteria commonly present in the environment, are emerging human pathogens associated with various infectious diseases in both immunocompetent and immunocompromised humans (1–15). Despite the increasing evidence of Aeromonas’ association with human infection and disease, Aeromonas spp. have only recently been recognized and confirmed as independent human pathogens (4, 6–10). Historical ambiguity in defining Aeromonas spp. as human pathogens (10, 16, 17) has resulted in a significant gap in knowledge regarding Aeromonas virulence and pathogenesis in humans. This ambiguity stemmed in part from the lack of routine speciation of Aeromonas isolates in clinical settings and surveillance studies, as Aeromonas cannot be reliably speciated with 16S-rRNA sequencing or standard microbiological and biochemical assays (6, 16, 18–21). This lack of species-level resolution precludes the ability to more comprehensively assess which species are contributing to human infection and virulence across species. This highlights the need to understand both the species relevant to human infection and Aeromonas species-specific virulence and pathogenesis in the context of human infection.
In the Global Enteric Multicenter Study (GEMS) (4, 11), a large case-control study surveying the burden of pathogens in pediatric diarrheal disease in Sub-Saharan Africa and Asia, Aeromonas spp. was among the top six enteric pathogens most commonly isolated from diarrheal cases in each age group examined and was isolated in >22% of diarrheal cases in Pakistan and Bangladesh (4). However, GEMS Aeromonas isolates were not speciated in real-time. Further studies by our group utilized genome sequencing and phylogenetic comparison to speciate a subset of GEMS Aeromonas spp. isolates (n = 431) (22). These analyses identified Aeromonas caviae as both the most prevalent species isolated, accounting for >50% of all isolates, and the only species significantly associated with human pediatric diarrheal disease (22). A recent study by Fernández-Bravo and Figueras identified that ~95% of human Aeromonas infections are attributed to only four Aeromonas species, A. caviae, A. dhakensis, A. veronii, and A. hydrophila, and A. caviae was most prevalent, accounting for 37.3% of infections (6). Augmented by the enhanced resolution of whole-genome sequencing and molecular methods, additional studies have recently established the prevalence of A. caviae in human Aeromonas infections (5, 23). In a multicenter cohort study of Aeromonas infections in adults in Japan, hepatobiliary disease was the most common *Aeromonas-*associated pathology, and A. caviae was the most abundant species, with isolation in 60% of cases (87/144) (5). Additionally, in a non-age restricted study of diarrheal cases in Northern Italy, Aeromonas was the second leading cause of bacterial diarrheal infection, accounting for 24.6% of cases, and A. caviae was isolated from 75% of the Aeromonas diarrheal cases (30/40) (23). Taken together, these studies form burgeoning evidence that A. caviae is a significant human pathogen and demonstrate the significant burden of A. caviae in human Aeromonas infections.
Despite these findings, and other evidence of A. caviae pathogenicity (15, 24) and burden in human Aeromonas infections (2), the pathogenicity of A. caviae has been debated (15, 25, 26). A. caviae are known to lack aerolysin, a T3SS, and heat-stable enterotoxin (ast), which are virulence factors commonly present in the other clinically relevant human-associated Aeromonas species (12, 25–29). The lack of these virulence factors resulted in the assumption that A. caviae was non-pathogenic (26). These assumptions have resulted in a systematic lack of understanding of A. caviae virulence in the human host, highlighting the urgent need for identification and characterization of A. caviae virulence factors.
In this study, we utilized comparative genomic analyses in >400 human-associated clinical GEMS Aeromonas spp. isolates (22) to identify genomic features specific to A. caviae with a putative role in virulence. We selected one representative putative A. caviae-specific virulence factor, flgB, predicted to encode a rod protein of the polar flagellar machinery and assembly apparatus (30–33), to functionally characterize in virulence and host–pathogen interactions. Given the role of A. caviae polar flagella in motility and adherence to human cell lines in limited previous studies (34–39), we hypothesized that deletion of flgB would affect virulence and host–pathogen interactions. To test our hypothesis, we genetically deleted flgB in a representative GEMS A. caviae isolate. Deletion of flgB abolished swimming motility and polar flagella assembly, decreased bacterial adherence to and proinflammatory cytokine production by human intestinal epithelial cell lines, as well as modestly increased survival in a Galleria mellonella larval in vivo model. These studies highlight the ability of this framework to identify and characterize A. caviae-specific genomic features that impact virulence and host–pathogen interactions.
RESULTS
Identification and mutagenesis of A. caviae-specific putative virulence factor, flgB
Mounting evidence of A. caviae’s prevalence in human infection (5, 6, 23, 28, 40) indicates A. caviae virulence, yet the virulence factors of human-associated A. caviae are not well characterized (27, 29). Previously, we conducted genome sequencing and phylogenetic comparison to speciate a representative subset of clinical human-associated Aeromonas isolates (n = 431) from GEMS (4, 11, 22). This analysis identified eight species: A. caviae (n = 230), A. veronii (n = 124), A. enteropelogenes (n = 29), A. dhakensis (n = 21), A. jandaei (n = 14), A. taiwanensis (n = 8), A. sanarellii (n = 4), and A. hydrophila (n = 1). Notably, A. caviae was the most prevalent species isolated (53.4%, 230/431) and the only species significantly associated with diarrheal disease (P = 0.004) compared to non-diarrheal controls (22).
We conducted comparative genomic analyses among these 431 Aeromonas genomes, utilizing the Roary (41) and Scoary (42) pipelines to compare A. caviae genomes (n = 230) to the genomes of the seven other Aeromonas species (n = 201). These analyses identified >300 genes that were significantly more abundant in A. caviae isolates than in the isolates of the other seven species (P < 0.0005). From these genes, we identified putative A. caviae-specific virulence factors as genes present in ≥90% of A. caviae isolates and ≤10% of other Aeromonas species isolates predicted to encode virulence factors in other enteric pathogens, such as genes involved in bacterial motility and adherence (8, 31, 34, 35). One such putative virulence gene was an flgB gene variant, which was significantly over-represented in A. caviae isolates (229/230 [99.6%]) compared to the isolates of the other seven Aeromonas species (12/201 [5.9%]) (P < 0.0005). Phylogenetic comparison of flgB nucleotide sequence across the 431 isolates confirmed the presence of an flgB gene in all Aeromonas species isolates. This analysis also confirmed the species specificity of flgB (Fig. S1A). The inferred flgB phylogenetic tree separates each species into distinct clades, with the exception being the flgB of A. taiwanensis and A. sanarellii isolates, which group with a subset of A. caviae isolates in a sub-clade (dark blue) nested within the A. caviae clade (light blue) (Fig. S1A). Allelic variation of flgB nucleotide sequence for species-specific variants (aligned to the flgB of a representative A. caviae) was observed as follows: A. caviae (7.9%), A. sanarellii (7.9%), A. taiwanensis (8.9%), A. hydrophila (15.1%), A. dhakensis (15.3%), A. veronii (22.5%), A. jandaei (19.3%), and A. enteropelogenes (19.5%) (Fig. S1B). Comparison of amino acid identity of the flgB from all Aeromonas species (aligned to the flgB of a representative A. caviae) ranged as follows: A. caviae (95.5%–100%), A. sanarellii (95.5%), A. taiwanensis (95.5%–96.2%), A. hydrophila (92.4%), A. dhakensis (90.9%–91.7%), A. veronii (88.6%–90.2%), A. jandaei (89.5%–90.2%), and A. enteropelogenes (90.2%–90.9%) (Fig. S1B). Amino acid similarity across apparent species-specific flgB variants ranged as follows: A. caviae (97%–100%), A. sanarellii (97%), A. taiwanensis (97%–98%), A. hydrophila (97%), A. dhakensis (95%–96%), A. veronii (93%–95%), A. jandaei (95%), and A. enteropelogenes (95%) (Fig. S1B). Nucleotide and amino acid sequence alignments for representative species-specific flgB variants are presented in Fig. S1C and D, respectively.
The flgB was predicted to encode a rod protein of the polar flagella basal body (30, 31), a component of the machinery involved in polar flagella assembly (32, 33). As A. caviae polar flagella (37) have been shown to confer swimming motility (34–36) and mediate adherence to some examined human cell lines (36–39), we hypothesized a putative role for flgB in A. caviae virulence. We selected this *A. caviae-*specific flgB as a representative putative virulence factor for mutagenesis and characterization in virulence and host–pathogen interactions, as there are functional assays with defined phenotypes to assess flagella functionally.
To assess the role of flgB in bacterial virulence and host–pathogen interactions, we generated an isogenic flgB deletion mutant (designated ΔflgB) in a representative human-associated A. caviae isolate from GEMS, 600239, utilizing the Lambda Red system (43, 44) (Table 1). We also generated two plasmid expression constructs for in trans complementation of our deletion mutant (Table 1). In the first construct (pBAH001), flgB is expressed from the Escherichia coli ompC promoter (PompC), which can be modulated by the osmolarity of the culture conditions (45, 46). To attempt to replicate endogenous regulation and flgB expression from the native promoter, the second construct (pBAH002) was engineered to contain flgB and 500 base pairs of sequence upstream of flgB, presumably containing the native promoter. Whole-genome sequencing confirmed definitive flgB deletion, with no other genetic effects, and successful introduction of complementation constructs in respective strains [designated ΔflgB (pBAH001) and ΔflgB (pBAH002)]. Bacterial fitness was not affected by flgB deletion nor complementation with either construct as compared to wild-type 600239, as determined by conducting growth curves in respective nutrient-rich media (Fig. S2).
Deletion of flgB abrogates aqueous motility and polar flagella production
As flgB is predicted to encode a component of the polar flagellar machinery (30–33), we utilized soft agar assays to assess swimming motility. Motility of ΔflgB (pBAH001) was assessed when culturing with 3 g/L NaCl (designated PompC low) or 10 g/L NaCl (designated PompC high), as expression from PompC can be modulated by osmolarity (45, 46). A representative image of the motility of all strains in soft agar plates ~22 h post-inoculation is shown in Fig. 1A. The ΔflgB mutant exhibited abrogated swimming motility compared to wild-type 600239 (P < 0.0001) (Fig. 1B). Motility of ΔflgB (pBAH001) (PompC low), ΔflgB (pBAH001) (PompC high), and ΔflgB (pBAH002) was restored to wild-type levels (P = 0.9430, 0.6920, and 0.7305, respectively) and significantly increased compared to that of ΔflgB (P < 0.0001, for all) (Fig. 1B), demonstrating functional complementation of swimming motility with both constructs. The motility of ΔflgB (pBAH001) (PompC low) was not significantly different than that of ΔflgB (pBAH001) (PompC high) (P = 0.4210). Therefore, we conducted all subsequent experiments culturing ΔflgB (pBAH001) with 10 g/L NaCl (PompC high).
Swimming motility of A. caviae strains by soft agar assay. The wild-type 600239, ΔflgB mutant, and complemented strains, ΔflgB (pBAH001) [flgB expression from the osmoregulated E. coli ompC promoter (PompC)] and ΔflgB (pBAH002) (expression from the native promoter), were inoculated into soft agar plates (0.25% agar) and incubated overnight at 37°C. The ΔflgB (pBAH001) strain was cultured in media containing 3 g/L NaCl (annotated PompC low) or 10 g/L NaCl (annotated PompC high) to examine motility when modulating expression from PompC. A representative image of the motility of each strain in soft agar plates is shown in panel A. Migration from the inoculation point was measured ~22 h post-inoculation (diameter, in mm) (B). Groups were compared by ordinary one-way ANOVA with Tukey’s test for multiple comparisons (**, P ≤ 0.0001). Data pooled from three independent experiments is shown (B).
To examine if the loss of motility was due to defects in polar flagella assembly, we visually assessed polar flagella production in the 600239 (upper left panel), ΔflgB (upper right panel), ΔflgB (pBAH001) (lower left panel), and ΔflgB (pBAH002) (lower right panel) strains using negative staining and transmission electron microscopy (Fig. 2). Polar flagella were present in the 600239 wild type; however, no polar flagella were observed in the ΔflgB mutant, indicating deletion of flgB abolished polar flagella assembly. Polar flagella were observed in both the ΔflgB (pBAH001) and ΔflgB (pBAH002) strains, indicating polar flagella assembly was functionally complemented with both constructs.
Visualization of polar flagella production in A. caviae strains by negative staining and transmission electron microscopy. The wild-type 600239 (upper left), ΔflgB mutant (upper right), and complemented strains, ΔflgB (pBAH001) (lower left) and ΔflgB (pBAH002) (lower right), were mounted on glow-discharged, formvar carbon-stabilized grids, stained with phosphotungstic acid, and visualized with transmission electron microscopy. Representative images are shown. Polar flagella are marked with arrows and scale bars indicate size (in μm).
Bacterial adherence and proinflammatory cytokine production were altered with flgB deletion
Previous studies have demonstrated A. caviae polar flagella mediate adherence to non-intestinal human cell lines (36–39, 48); however, there are limited studies assessing A. caviae adherence and other host–pathogen interactions in relevant human intestinal cell lines (23, 48–50). Additionally, the location of A. caviae binding and infection in the human gastrointestinal tract remains unknown (51). We utilized relevant HT-29 and Caco2 human intestinal epithelial cell lines, representative of colonic cells (52) and small intestinal epithelial cells (38, 52), respectively, to characterize the effects of the flgB mutation on bacterial adherence and proinflammatory cytokine production.
In the HT-29 cells, ΔflgB exhibited significantly decreased adherence compared to wild-type 600239 (P < 0.0001) (Fig. 3A). Adherence of ΔflgB (pBAH001) and ΔflgB (pBAH002) was not significantly different compared to that of ΔflgB (P = 0.3623 and 0.7104, respectively) (Fig. 3A), indicating complementation with either pBAH001 or pBAH002 did not rescue adherence in the HT-29 cells. In the Caco2 cells, flgB deletion had no effect on adherence, with similar adherence observed in the wild-type and ΔflgB mutant (P = 0.9794) (Fig. 3B). Adherence of ΔflgB (pBAH001) and ΔflgB (pBAH002) was not significantly different than that of the wild-type 600239 (P = 0.8981 and 0.5022, respectively), or ΔflgB mutant (P = 0.7003 and 0.2914, respectively) (Fig. 3B).
Adherence of A. caviae strains in HT-29 and Caco2 intestinal epithelial cell lines. Confluent monolayers of HT-29 (A) and Caco2 (B) cells were infected with the wild-type 600239, ΔflgB mutant, or complemented strains, ΔflgB (pBAH001) or ΔflgB (pBAH002) (in technical triplicate) at MOI = 0.05. Fresh Dulbecco’s Modified Eagle Medium was used as a negative control. Four hours post-infection, monolayers were washed to remove non-adhered planktonic bacteria, lysed with Triton X, and the resulting mixture was serially diluted and plated for enumeration of CFU of adhered bacteria. Groups were compared by ordinary one-way ANOVA with Tukey’s test for multiple comparisons (**, P ≤ 0.0001; ns, non-significant); adherence was not significantly different between any of the experimental groups in the Caco2 cell line (B). Data presented is pooled from three independent experiments.
Production of proinflammatory cytokines interferon-gamma (IFN-γ), tumor necrosis factor-alpha (TNF-α), IL-1β, IL-2, IL-4, IL-6, IL-8, IL-10, IL-12p70, and IL-13 was measured by multiplex cytokine assay using spent media supernatants from the adherence assays in both HT-29 (Fig. 4A through E) and Caco2 (Fig. 5A through E) cells. TNF-α, IL-2, IL-4, IL-10, and IL-12p70 were not produced at 4 h post-infection with any of the bacterial strains, in either HT-29 or Caco2 cell lines (data not shown). Induction of IL-8 (Fig. 4A), IL-13 (Fig. 4B), IFN-γ (Fig. 4C), IL-1β (Fig. 4D), and IL-6 (Fig. 4E) was significantly reduced when infecting HT-29 cells with the ΔflgB mutant compared to the 600239 wild type (P < 0.0001 for all). Complementation with pBAH001 or pBAH002 partially rescued production of IL-8 (Fig. 4A), IL-13 (Fig. 4B), IFN-γ (Fig. 4C), IL-1β (Fig. 4D), and IL-6 (Fig. 4E), with greater cytokine production occurring with ΔflgB (pBAH001) infection compared to ΔflgB (pBAH002). IL-8 (Fig. 5A), IL-13 (Fig. 5B), IL-1β (Fig. 5D), and IL-6 (Fig. 5E) production was significantly decreased in Caco2 cells infected with the ΔflgB mutant compared to 600239 wild type (P < 0.0001 for all). IFN-γ production did not differ significantly between any strains in the Caco2 infections (Fig. 5C). In contrast to observations in the HT-29 cells, there was no restoration of IL-8 (Fig. 5A), IL-13 (Fig. 5B), IL-1β (Fig. 5D), or IL-6 (Fig. 5E) production when infecting Caco2 cells with ΔflgB (pBAH001) or ΔflgB (pBAH002), indicating a lack of functional complementation with either construct.
Proinflammatory cytokine production in HT-29 intestinal epithelial cell lines infected with A. caviae strains. Confluent monolayers of HT-29 cells were infected with the wild-type 600239, ΔflgB mutant, or complemented strains, ΔflgB (pBAH001) or ΔflgB (pBAH002) at MOI = 0.05 for 4 h. Each strain was assayed in technical triplicate and fresh Dulbecco’s Modified Eagle Medium was used as a negative control. A multiplex cytokine kit was used to measure the production of proinflammatory cytokines, including IL-8 (A), IL-13 (B), IFN-γ (C), IL-1β (D), and IL-6 (E), in spent media supernatants. Groups were compared by ordinary one-way ANOVA with Tukey’s test for multiple comparisons (***, ***, **, and , P ≤ 0.0001, P ≤ 0.001, P ≤ 0.01, and P ≤ 0.05, respectively). Data pooled from three independent experiments is shown.
Proinflammatory cytokine production in Caco2 intestinal epithelial cell lines infected with A. caviae strains. Confluent monolayers of Caco2 cells were infected with the wild-type 600239, ΔflgB mutant, or complemented strains, ΔflgB (pBAH001) or ΔflgB (pBAH002) at MOI = 0.05 for 4 h. Strains were assayed in technical triplicate and fresh Dulbecco’s Modified Eagle Medium was used as a negative control. A multiplex cytokine kit was used to measure the production of proinflammatory cytokines, including IL-8 (A), IL-13 (B), IFN-γ (C), IL-1β (D), and IL-6 (E), in spent media supernatants. Groups were compared by ordinary one-way ANOVA with Tukey’s test for multiple comparisons (***, ***, **, and , P ≤ 0.0001, P ≤ 0.001, P ≤ 0.01, and P ≤ 0.05, respectively); (ns, non-significant). Data pooled from three independent experiments is shown.
Examination of the role of flgB in a G. mellonella survival model
Small animal models are often not suitable for in vivo assessment of enteric pathogens, including Aeromonas, as infection does not result in diarrhea, the main symptom of enteric infection in humans (25, 53, 54). Therefore, we assessed in vivo virulence in a G. mellonella larval model. Larvae were injected with 10^6^, 10^5^, or 10^4^ CFU of each strain and survival was monitored for 72 h. Survival between experimental groups was compared by the Log-rank (Mantel-Cox) test. All four A. caviae strains exhibited a dose response, and survival was significantly increased when infecting with 10^4^ CFU vs 10^5^ CFU (P < 0.0001), and with 10^5^ CFU vs 10^6^ CFU (P < 0.0001) (Fig. 6A; Fig. S3, Table S2). Infections with 10^6^ CFU yielded <2% survival rates in all experimental groups by 22 h post-infection, precluding survival comparisons at this dose (Table S2). In contrast, when infectedg with 10^4^ CFU, survival rates in all four experimental groups remained >56% and never decreased below the threshold of 50% survival (Table S2). Therefore, we assessed survival with 10^5^ CFU, which yielded an effective range of survival to measure the impact of genetic changes (Fig. 6A; Fig. S3, Table S2).
In vivo virulence of A. caviae strains in the G. mellonella larvae model. To assess dose response, G. mellonella larvae (n = 20–24 per group) were infected with 106, 105, or 104 CFU of the wild-type 600239 strain (A). To assess virulence across strains, G. mellonella larvae (n = 20–24 per group) were infected with 105 CFU of the 600239, ΔflgB mutant, or complemented strains, ΔflgB (pBAH001) or ΔflgB (pBAH002) (B). Larvae were infected via injection into the hemolymph. Controls were injected with an equal volume of inoculum diluent. Larvae were maintained at 37°C and survival was assessed at 22, 48, and 72 h post-infection. Survival curves were individually compared between each experimental group using the Log-rank (Mantel-Cox) test and data pooled from three independent experiments are shown (**, P ≤ 0.0001; ns, non-significant). Survival was not significantly different between any of the experimental groups infected with 105 CFU.
Survival was greater with ΔflgB compared to wild-type 600239 at each infection time point. Although not statistically significant, the overall difference in survival rates between the wild type and mutant is trending toward significance (P = 0.0958) (Fig. 6B; Table S2). Survival in ΔflgB (pBAH001) and ΔflgB (pBAH002) infection was not significantly different compared to wild-type or ΔflgB infection (Table S2).
DISCUSSION
There is significant and expanding evidence that A. caviae is an independent human pathogen associated with numerous clinical pathologies (5, 6, 12, 22, 23, 28, 40). Recent studies demonstrate that while four Aeromonas species account for 95% of all human Aeromonas infections, A. caviae is responsible for the most significant burden (6). The prevalence of A. caviae compared to other Aeromonas species, across various infectious diseases, geographic locations, age cohorts, and socioeconomic demographics highlights A. caviae virulence (5, 6, 22, 23). However, historical uncertainty in identifying A. caviae as a human pathogen, as well as barriers to reliable speciation of Aeromonas precluding routine speciation of Aeromonas isolates in surveillance studies and clinical settings, has contributed to the lack of characterization of virulence factors specific to A. caviae (16, 17, 54). In the current study, we conducted comprehensive comparative genomic analyses (41, 42) in a large sample set of human Aeromonas isolates from GEMS (n = 431) (11, 22) and identified an flgB gene variant as a representative putative A. caviae-specific virulence factor. This flgB variant was significantly more prevalent among A. caviae isolates than in the other Aeromonas species and was predicted to encode a polar flagellar basal body protein (30, 31) putatively involved in flagellar assembly (32, 33). Notably, our phylogenetic analysis of the flgB gene across the 431 GEMS Aeromonas isolates, encompassing eight species, confirmed species specificity of flgB, with the exception of the flgB of A. sanarellii and A. taiwanensis. This data aligned with our comparative genomic analysis, wherein the A. taiwanensis (n = 4) and A. sanarellii (n = 8) isolates are 12 of the 201 (5.9%) non-A. caviae Aeromonas species isolates identified by the comparative genomic analysis to contain a flgB gene similar to the A. caviae-specific flgB variant. To note, it is unknown if the changes across species-specific flgB variants result in functional or antigenic variation. We hypothesized a putative role for flgB in virulence as the A. caviae polar flagella (37) have been demonstrated to mediate swimming motility (34–36) and host-cell adherence (36–39), both of which are important tenants of virulence for enteric pathogens (8, 31, 34, 35).
Our large-scale comparative genomic analyses conducted in reliably speciated isolates are important advances in assessing A. caviae-specific virulence factors. Previous studies of A. caviae virulence factors have been limited, both in number and in scope (12, 27, 29, 55, 56). Numerous studies have characterized the virulence of Aeromonas isolates by screening for the presence/absence of a limited number of previously characterized virulence genes commonly present in other human-associated Aeromonas species isolates (12, 27, 55, 56). This approach is limited as it perpetuates a static understanding of Aeromonas virulence factors, minimizes the discovery of new virulence factors, and operates on the assumption that those limited virulence factors are determinants of virulence for all Aeromonas species. Notably, A. caviae lacks several common Aeromonas virulence factors included in the virulence screens, such as aerolysin, a T3SS, and heat-stable enterotoxin (ast), which resulted in historic assumptions that A. caviae was non-pathogenic (12, 25–29). However, the accumulating epidemiological data paint a very different picture, wherein A. caviae is emerging as one of the most prevalent Aeromonas species underlying human Aeromonas infections and pathologies (5, 6, 12, 23, 28, 40). Indeed, Chong et al. recently conducted an expansive analysis characterizing virulence genes in 565 A. caviae isolates, derived from both intestinal and extraintestinal human infections, as well as zoonotic and environmental sources (29). While this study is exciting and contributes to the characterization of A. caviae virulence genes, this work focused solely on A. caviae and did not include any other Aeromonas species in the analysis, thereby precluding the ability to assess differences in virulence gene content across species. In the current study, we applied extensive genomic comparisons, using a large sample set of relevant, reliably speciated clinical isolates, with inter-species comparisons, to systematically assess A. caviae-specific virulence factors and ultimately identified flgB as a putative A. caviae-specific virulence factor. A. caviae-specific virulence factors remain poorly characterized (27, 29), highlighting the significance of this work in developing a platform to identify and characterize virulence factors specific to A. caviae.
By generating an isogenic flgB deletion mutant and two distinct plasmid complementation constructs (Table 1), we developed a system to examine the role of a representative putative A. caviae-specific virulence factor, flgB, in aspects of bacterial virulence (motility and polar flagella production) and host–pathogen interactions (adherence, proinflammatory cytokine production, and in vivo virulence). We establish the essential role of A. caviae flgB in polar flagellar assembly and swimming motility ([Fig. 1 and 2](#F1 F2)). Swimming motility and polar flagella assembly were abolished in the mutant and functionally complemented with expression from both the osmoregulated E. coli PompC and the native promoter, demonstrating flgB is required for motility and polar flagellar assembly in A. caviae.
The host–pathogen interactions of A. caviae in human gastrointestinal infection remain understudied, and notably, it is unknown where A. caviae infects the human gastrointestinal tract (51). To address this, we examined the effects of flgB deletion on host–pathogen interactions, specifically adherence and proinflammatory cytokine production, in HT-29 and Caco2 human intestinal epithelial cell lines, representative of colonic cells and small intestinal epithelial cells, respectively (38, 52). In line with previous studies characterizing A. caviae adherence in human cell lines (23, 36–39, 48–50), we observed adherence of our A. caviae strains to both human intestinal cell lines. Interestingly, adherence of the wild-type A. caviae was greater in the HT-29 cells compared to Caco2 cells. While deletion of flgB significantly decreased adherence to HT-29 cells compared to the wild type (Fig. 3A), adherence was not rescued by complementation (Fig. 3A). In contrast, adherence to the Caco2 cells was similar across wild-type, mutant, and complemented strains (Fig. 3B). These data suggest the A. caviae polar flagella plays a role in mediating adherence to HT-29 cells but is not essential for adherence to Caco2 cells. This indicates other factors may be involved in adherence to these cells, in addition to the polar flagella. To characterize the host response to A. caviae infection, we examined proinflammatory cytokine production. We observed significant induction of IL-8, IL-13, IL-1β, and IL-6 in both HT-29 and Caco2 cell lines with wild-type 600239 infection (Fig. 4A through E and 5A through E); IFN-γ was significantly induced with wild-type infection in the HT-29 cells only (Fig. 4C). These data corroborate a previous study documenting IL-8 production in in vitro infection with clinical A. caviae isolates (23). Additionally, production of IL-8, IL-1β, and IL-6 is known to be induced by TLR5 recognition of flagellin (8, 57), the polar flagella structural subunit, and IFN-γ has been reported to modulate an inflammatory response to flagellin in the gut (58). As functional polar flagella are not assembled in our flgB mutant (Fig. 2), we anticipate a lack of surface-exposed flagellin in the flgB mutant. Our findings align with this as flgB deletion significantly decreased production of IL-8, IL-13, IL-1β, and IL-6 in both cell lines, and of IFN-γ in the HT-29 cells. Overall, these results demonstrate that the A. caviae polar flagella are involved with eliciting a proinflammatory response to A. caviae in both human intestinal cell lines. Additionally, the cumulative differences observed in adherence and cytokine production between the HT-29 and Caco2 cell lines could indicate possible “tropism” of A. caviae within the gastrointestinal tract. This tropism will need to be examined in human intestinal enteroid models, which are developed from human intestinal biopsies from various sections of the gastrointestinal tract. Therefore, these models contain greater cellular complexity than immortalized cell lines and are representative of the colon and various sections of the small intestine (59).
In vivo models are critical for investigating host–pathogen interactions of human pathogens. However, in vivo assessment of enteric pathogens is significantly limited due to a lack of relevant small animal models, as diarrhea, the hallmark phenotype of human enteric infections, is often not produced in these models (25, 53, 54). Mouse models can be utilized as a survival-based virulence screening platform; however, in limited studies, infection with human diarrheal A. caviae isolates yielded LD_50_ values of 2–6 × 10^9^ CFU (25, 54). The G. mellonella larval model is a survival-based virulence screening platform that lacks the inherent hurdles of murine models (53, 60), thereby facilitating cost-effective interrogation of greater numbers of organisms, translating to more powered assessments, without the need for regulatory approval, skilled technical training, or specialized personnel and facilities. This model has been used to assess the virulence of other enteric pathogens, Shigella spp., C. jejuni, and Y. enterocolitica, thus confirming this model as a valuable screening platform for enteric pathogens (53, 60). The G. mellonella model was previously utilized in a single study to assess the virulence of environmental A. hydrophila, veronii, and salmonicida isolates. However, our current study will be the first application of this model for assessing the virulence of A. caviae, and of a human-associated clinical Aeromonas isolate (61). We achieved consistent and reproducible dose responses for all A. caviae strains, demonstrating the suitability of the model to assess A. caviae virulence in vivo (Fig. 6 and Table S2). Deletion of flgB modestly, but not significantly, increased survival compared to the wild type. This data suggests A. caviae pathogenesis in G. mellonella most likely involves additional factors.
In conclusion, we leveraged the power of comparative genomics and a large sample set of accurately speciated human clinical Aeromonas isolates (n = 431) to identify genetic factors specific to A. caviae with putative roles in virulence. We characterized one representative putative *A. caviae-*specific virulence factor, flgB, and demonstrated its essential role in aqueous motility and polar flagellar formation using a deletion mutant and complementation constructs generated in this study. Deletion of flgB significantly decreased bacterial adherence to HT-29 cells and interrupted the production of proinflammatory cytokines in both HT-29 and Caco2 human intestinal cells. Lastly, we characterized in vivo virulence of A. caviae in the G. mellonella larval survival model for the first time and observed a non-significant increase in survival with flgB deletion compared to the wild type in this model. Taken together, these data suggest a role for flgB in bacterial virulence and host–pathogen interactions. Lastly, we established a framework for the identification and characterization of A. caviae-specific virulence factors, coupling comparative genomic analyses with the genetic systems, functional assays, and the in vivo and in vitro models we optimized here.
MATERIALS AND METHODS
Bacterial strains, culture conditions, and reagents
Bacterial strains utilized in this study are listed in Table 1. Strains were cultured from frozen stocks in Mueller-Hinton medium (BD) [for 600239, ΔflgB, and ΔflgB (pBAH002)] or Lysogeny broth (LB) medium (MP Biomedicals) [for ΔflgB (pBAH001)] and incubated overnight at 37°C unless otherwise stated. For antibiotic selection, 50 μg/mL neomycin (Neo50) (Sigma) or 20 μg/mL chloramphenicol (Cm20) (Sigma) was added to culture media when applicable. The ΔflgB (pBAH001) strain was cultured in LB containing 3 g/L NaCl or 10 g/L NaCl, as expression from the E. coli ompC promoter (PompC) can be increased by increasing the osmolarity of culture conditions (45, 46). However, the regulation of PompC is not significantly stringent enough to eliminate gene expression. Molecular biology techniques were performed according to standard protocols (62). GoTaq Green DNA polymerase master mix (Promega) or PfuUltra II Fusion HS DNA Polymerase (Agilent) was utilized for PCR amplifications. Restriction enzymes and T4 DNA ligase were acquired from NEBioLabs. Primers were generated by Integrated DNA Technologies. All primers and PCR amplification conditions are listed in Table S1. Plasmids and plasmid constructs are listed in Table 1.
Comparative genomics
For all comparative genomic analyses, we utilized draft genomes we previously generated for a representative set of 431 clinical human-associated Aeromonas isolates collected in the GEMS (4, 11, 22). Sequencing, genome assembly, and phylogenetic comparison were conducted as previously described (22, 63). Eight species are represented in this sample set as determined by our prior phylogenetic comparison analysis: A. caviae (n = 230), A. veronii (n = 124), A. enteropelogenes (n = 29), A. dhakensis (n = 21), A. jandaei (n = 14), A. taiwanensis (n = 8), A. sanarellii (n = 4), and A. hydrophila (n = 1). Assembled genomes were annotated with Prokka (version 1.14.6) (64), and the Generated General Feature Formats were analyzed using Roary (version 3.13.0) (41). A. caviae genomes (n = 230) were compared to the genomes of the other seven Aeromonas species identified in GEMS (n = 201) using Scoary (version 1.6.16) (42). Results were evaluated with Bonferroni’s method for multiple comparisons within Scoary analysis (42). To identify *A. caviae-*specific putative virulence factors, we first categorized *A. caviae-*specific genes as those significantly more abundant in A. caviae isolates than in the isolates of the other seven species (P < 0.0005), and present in ≥90% of A. caviae isolates and in ≤10% of other Aeromonas species isolates. We manually assessed the A. caviae-specific genes and identified putative virulence factors as genes predicted to encode virulence factors in other enteric pathogens, such as genes involved in bacterial motility and adherence (8, 31, 34, 35). An A. caviae-specific flgB gene variant, predicted to encode a polar flagella basal body rod protein (30, 31), was selected as a representative putative virulence factor, as polar flagella can be assessed with well-defined functional assays.
Phylogenetic analysis of flgB
BLASTN (65) and MUSCLE (66) were used to align the flgB nucleotide sequence across the 431 Aeromonas isolates to assess divergence. IQ-TREE (v2) was utilized to infer a midpoint-rooted maximum-likelihood phylogeny (67). The inferred phylogeny was visualized with FigTree (v 1.4.4) (https://tree.bio.ed.ac.uk/software/figtree/). Nucleotide sequences of flgB across the 431 Aeromonas isolates were translated (68), and BLASTP (65) was utilized to assess amino acid identity and similarity. Nucleotide and amino acid sequences of representative species-specific flgB variants were aligned using CLUSTALW (version 2.1) (69) using default parameters to generate the alignments for Fig. S1.
Lambda Red mutagenesis
Stable chromosomal flgB deletion was carried out in a single, representative clinical GEMS A. caviae isolate 600239 utilizing the Lambda Red recombineering system as previously described (43, 44). Briefly, plasmid pKM200 (44) was introduced into the 600239 strain by electroporation; transformants were selected on LB+Cm20 at 30°C and screened for the pKM200 plasmid via PCR using primer set JMM01-F/R (Table S1). Expression of the Lambda Red recombinase was induced with 1 mM IPTG, then 600239 was heat-shocked at 42°C for 15 min and made electrocompetent (44).
Using primer set BAH01-F/R (Table S1), 66–67 bp of the sequence flanking flgB were PCR amplified onto a kanamycin/neomycin resistance cassette (aph) from helper plasmid, pKD4 (43). The ~1.6 kb product was electroporated into 600239 expressing the Lambda Red recombinase, plated on MH+Neo50, and neomycin-resistant isolates were screened by PCR for the ΔflgB::aph mutation using primer set BAH02-F/R (Table S1). The aph cassette was removed from the genome at FRT sites by an FLP flippase carried on the pCP20 plasmid (43). pCP20 was introduced into 600239 ΔflgB by electroporation and selected on LB+Cm20 at 30°C. Flippase activity was induced by incubation at 42°C, and transformants were screened for loss of phenotypic resistance (43) by patching on MH and counterselection on MH+Neo50. Loss of the aph cassette was verified by PCR in resultant clones using primer set BAH02-F/R (Table S1). Whole-genome sequencing (SeqCenter) was utilized to confirm flgB deletion, removal of the aph cassette, and absence of other mutations/genetic effects of the molecular engineering.
Generation of expression constructs for complementation
Plasmid expression constructs for trans complementation of flgB were generated in pSEC10 (45, 46) for flgB expression from the osmoregulated E. coli ompC promoter (PompC) (pBAH001) and pSEC10-M (47) for expression from the native promoter (pBAH002) (Table 1). To generate pBAH001, flgB was PCR amplified from 600239, with 5′ BamHI and 3′ NheI restriction sites, using PfuUltra II Fusion HS DNA Polymerase (Agilent), CleanAmp 7-deaza-dGTP Mix (TriLink), and primer set BAH03-F/R (Table S1). The resulting 420 bp product was purified by phenol-chloroform extraction, precipitated with 125 mM NaCl and ethanol, rescued in the pCR-Blunt II-TOPO vector (Invitrogen) using the Zero Blunt TOPO PCR Cloning Kit (Invitrogen), transformed into One Shot TOP10 Chemically Competent E. coli (Invitrogen) per manufacturer’s instructions, and selected on MH+Neo50. The resulting construct, pCR-Blunt II-TOPO::flgB (Table 1), was confirmed by restriction enzyme digest and Sanger Sequencing (GENEWIZ). Plasmid pSEC10 (45, 46) was digested with BamHI and NheI, and a ~6.3 kb fragment was excised following agarose gel electrophoresis and gel purified (Invitrogen PureLink Quick Gel Extraction and PCR Purification Combo Kit). pCR-Blunt II-TOPO::flgB was digested with BamHI and NheI, separated by agarose gel electrophoresis, and a 420 bp fragment was excised, electroeluted, and purified by phenol-chloroform extraction. The purified 420 bp fragment was ligated into the BamHI/NheI site of pSEC10, downstream of PompC, transformed into TOP10 E. coli as above*,* selected on MH+Neo50, and PCR-screened using primer set BAH03-F/R (Table S1), yielding pBAH001 (Table 1).
To generate pBAH002, flgB, along with 500 bp of upstream sequence, was PCR amplified from 600239, with 5′ NheI and 3′ AvrII restriction sites, using PfuUltra II Fusion HS DNA Polymerase (Agilent), CleanAmp 7-deaza-dGTP Mix (TriLink), and primer set BAH04-F/R (Table S1). The 920 bp product was purified by phenol-chloroform extraction, precipitated with 125 mM NaCl and ethanol, rescued in pCR-Blunt II-TOPO (Invitrogen), transformed into TOP10 E. coli as above, and selected on MH+Neo50. The resulting construct, pCR-Blunt II-TOPO::flgB+500 (Table 1), was confirmed by restriction enzyme digest and Sanger Sequencing (GENEWIZ). pCR-Blunt II-TOPO::flgB+500 was digested with NheI and AvrII, and a 920 bp fragment was electroeluted from an agarose gel fragment, purified by phenol-chloroform extraction, ligated into the NheI/AvrII site of plasmid pSEC10-M (47), transformed into TOP10 E. coli as above, selected on MH+Neo50, and PCR-screened using primer set BAH04-F/R (Table S1), yielding pBAH002 (Table 1). Purified pBAH001 and pBAH002 were separately electroporated into the ΔflgB 600239 strain, selected on MH+Neo50, and PCR-screened using primer sets BAH03-F/R and BAH04-F/R, respectively (Table S1). Introduction of pBAH001 and pBAH002 and the host genome sequence was confirmed by whole-genome sequencing (SeqCenter).
Growth curve assays
Overnight broth cultures of each strain (Table 1) were diluted 1:500 in fresh media and assayed in technical triplicate in 96-well, flat-bottom plates at 37°C with agitation. The OD_600_ was measured every 3 min for ~16.5 h using the Stratus (MRODX1r2) continuous plate reader (Cerillo). Fresh culture media served as baseline controls. Results were collated using the Cerillo Labrador software (v2.3.3). Two independent experiments were conducted.
Motility assays
Overnight broth cultures of each strain (Table 1) were inoculated into the center of soft agar plates (0.25% agar) with a sterile toothpick. Plates were incubated at 37°C (not inverted) and migration from the inoculation point (diameter) was measured ~22 h post-inoculation. Three independent experiments were performed.
Transmission electron microscopy
For all strains (Table 1), overnight cultures on agar plates were submitted to the University of Maryland, Baltimore Electron Microscopy Core for negative staining and transmission electron microscopy imaging as described previously (70). Single colonies were transferred to glow-discharged, formvar carbon-stabilized grids by floating the grid over a single colony in a droplet of filtered water to retain bacterial conformation and morphology. Grids were stained with 2% aqueous phosphotungstic acid, air-dried, and imaged in an FEI Tecnai T12 (Thermo Fisher) transmission electron microscope at 80 kV with an AMT bottom-mount camera.
Cell culture and adherence assays
HT-29 (ATCC HTB-38) and Caco2 (ATCC HTB-37) cell lines were cultured in 75 cm^2^ flasks at 37°C, 5% CO_2_ in Dulbecco’s Modified Eagle Medium (DMEM) (Gibco) supplemented with 10% and 20% heat-inactivated fetal bovine serum (FBS) (Gibco), respectively.
For adherence assays, cells were seeded into 12-well plates (Corning) and cultured to confluence. To prepare the inocula, overnight broth cultures of each strain (Table 1) were diluted in fresh media, cultured to 0.5 OD_600_, pelleted, and resuspended in an equal volume of DMEM(-FBS) (Gibco). Inocula were diluted to MOI = 0.05 in DMEM(-FBS). Inocula were serially diluted and plated for enumeration of CFU.
Confluent monolayers of HT-29 or Caco2 cells were infected with each strain in technical triplicate, at MOI = 0.05 for 4 h (37°C, 5% CO_2_). DMEM(-FBS) served as a negative control. Four hours post-infection, spent media supernatants were collected for cytokine production assays, then monolayers were washed three times with fresh DMEM(-FBS), lysed with Triton-X (ThermoFisher), serially diluted, and plated for enumeration of CFU of adhered bacteria. Three independent experiments were performed for both cell types.
Cytokine assays
Spent media supernatants from the adherence assays in HT-29 and Caco2 cells were collected 4 h post-infection, centrifuged (12,000 × g, 5 min, 4°C) to remove bacterial/cellular debris, and stored at −80°C until use. MSD U-PLEX Biomarker Group 1 (human) Multiplex Assays (K15049K-1) (Mesoscale Diagnostics [MSD]) were utilized to assess the production of 10 pro-inflammatory cytokines: interferon-gamma (IFN-γ), tumor necrosis factor-alpha (TNF-α), and interleukins IL-1β, IL-2, IL-4, IL-6, IL-8, IL-10, IL-12p70, and IL-13. Each sample was run in technical duplicate, and assays were conducted per the manufacturer’s instructions. Data were analyzed using the MSD Discovery Workbench (version 4.0; https://www.mesoscale.com/en/products_and_services/software).
G. mellonella survival assays
Larvae of the Greater Wax Moth, G. mellonella, were utilized as an alternative model for screening virulence in vivo as described previously (53, 71), with minor modifications. Briefly, G. mellonella larvae (Vanderhorst Wholesale; https://www.waxworms.net) weighing 210–300 mg, and showing no signs of disease (active, cream-colored), were sorted into Petri dishes with sawdust bedding one day prior to infection (n = 20–24 larvae per group); larvae were utilized within 2–3 days of receipt. Prior to injection, larvae were immobilized at 4°C (30–40 min), then incubated on ice immediately preceding injection. A group of non-injected larvae served as environmental controls.
For inocula preparation, overnight broth cultures of each strain (Table 1) were diluted 1:100 in fresh media, cultured to 0.5 OD_600_, pelleted, and resuspended in an equal volume of DMEM (Gibco). To assess dose response, inocula were diluted to 10^8^, 10^7^, or 10^6^ CFU/1 mL DMEM. Ten microliters of inoculum were injected into the hemolymph via injection into the last, right proleg using 1 cc insulin syringes (31G needle, 5/16) (BD). This translated to 10^6^, 10^5^, or 10^4^ CFU in the larvae, respectively. Controls were injected with an equal volume of inocula diluent, DMEM. Larvae were incubated at 37°C and survival was assessed at 22, 48, and 72 h post-infection. Larvae were deemed dead with failure to move upon prodding. Inocula were serially diluted and plated for enumeration of CFU. Three independent experiments were performed.
Statistics
GraphPad Prism (Version 10.4.1) was utilized to visualize data and conduct analysis for all statistical assessments, with the exception of those conducted within the Scoary pipeline (42). Data for motility, adherence, and cytokine production assays were assessed by ordinary one-way ANOVA with Tukey’s test for multiple comparisons; data are shown as mean ± standard deviation. Survival in the G. mellonella model was plotted in Kaplan-Meier curves, and the log-rank (Mantel-Cox) test was used to compare survival between two groups across all comparisons. Significance is indicated as follows: ****, ***, **, and *, P ≤ 0.0001, P ≤ 0.001, P ≤ 0.01, and P ≤ 0.05, respectively.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Alatorre-Fernández CP, Cornejo-Juárez P, Velázquez-Acosta C, Volkow-Fernández P. 2023. Bacteremia caused by Aeromonas species in patients with cancer: clinical manifestations and outcomes. J Infect Dev Ctries 17:359–366. doi:10.3855/jidc.1753037023428 · doi ↗ · pubmed ↗
- 2Albert MJ, Ansaruzzaman M, Talukder KA, Chopra AK, Kuhn I, Rahman M, Faruque AS, Islam MS, Sack RB, Mollby R. 2000. Prevalence of enterotoxin genes in Aeromonas spp. isolated from children with diarrhea, healthy controls, and the environment. J Clin Microbiol 38:3785–3790. doi:10.1128/JCM.38.10.3785-3790.200011015403 PMC 87476 · doi ↗ · pubmed ↗
- 3Igbinosa IH, Igumbor EU, Aghdasi F, Tom M, Okoh AI. 2012. Emerging Aeromonas species infections and their significance in public health. Scientific World Journal 2012:625023. doi:10.1100/2012/62502322701365 PMC 3373137 · doi ↗ · pubmed ↗
- 4Qamar FN, Nisar MI, Quadri F, Shakoor S, Sow SO, Nasrin D, Blackwelder WC, Wu Y, Farag T, Panchalingham S, et al.. 2016. Aeromonas-associated diarrhea in children under 5 years: the GEMS experience. Am J Trop Med Hyg 95:774–780. doi:10.4269/ajtmh.16-032127527635 PMC 5062620 · doi ↗ · pubmed ↗
- 5Sakurai A, Suzuki M, Ohkushi D, Harada S, Hosokawa N, Ishikawa K, Sakurai T, Ishihara T, Sasazawa H, Yamamoto T, Takehana K, Koyano S, Doi Y. 2023. Clinical features, genome epidemiology, and antimicrobial resistance profiles of Aeromonas spp. causing human infections: a multicenter prospective cohort study. Open Forum Infect Dis 10:ofad 587. doi:10.1093/ofid/ofad 58738156048 PMC 10753922 · doi ↗ · pubmed ↗
- 6Fernández-Bravo A, Figueras MJ. 2020. An update on the genus Aeromonas: taxonomy, epidemiology, and pathogenicity. Microorganisms 8:129. doi:10.3390/microorganisms 801012931963469 PMC 7022790 · doi ↗ · pubmed ↗
- 7Janda JM, Abbott SL. 2010. The genus Aeromonas: taxonomy, pathogenicity, and infection. Clin Microbiol Rev 23:35–73. doi:10.1128/CMR.00039-0920065325 PMC 2806660 · doi ↗ · pubmed ↗
- 8Tomás JM. 2012. The main Aeromonas pathogenic factors. ISRN Microbiol 2012:256261. doi:10.5402/2012/25626123724321 PMC 3658858 · doi ↗ · pubmed ↗