Acute Hypoxemic Respiratory Failure Caused by Nonspecific Interstitial Pneumonia in Mixed Connective Tissue Disease: A Case Report
Syed M Naqvi, Amin Ur Rehman Nadeem, Jason Liu, Tyler Paul, Sara R Oliveira

TL;DR
A 42-year-old man with acute lung failure was found to have a rare autoimmune condition causing lung disease, which improved with immunosuppressive treatment.
Contribution
This case report presents a rare instance of MCTD-associated NSIP causing acute respiratory failure and highlights its successful treatment.
Findings
The patient showed significant improvement after high-dose corticosteroids and mycophenolate mofetil.
MCTD-associated NSIP can present acutely with hypoxemic respiratory failure.
Early immunosuppressive therapy is effective in treating this condition.
Abstract
Mixed connective tissue disease (MCTD) is an autoimmune overlap condition characterized by features of systemic lupus erythematosus, systemic sclerosis, and polymyositis, frequently involving pulmonary manifestations. Interstitial lung disease (ILD) is one of its most serious complications and may present acutely with hypoxemic respiratory failure. Among the ILD patterns associated with MCTD, nonspecific interstitial pneumonia (NSIP) predominates and is characterized by diffuse ground-glass opacities, reticulation, and potential responsiveness to early immunosuppressive therapy. We describe a 42-year-old male active-duty service member who presented with acute hypoxemic respiratory failure and was ultimately diagnosed with MCTD-associated NSIP following bronchoscopy and confirmatory serologies. He demonstrated marked clinical and radiographic improvement after receiving high-dose…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
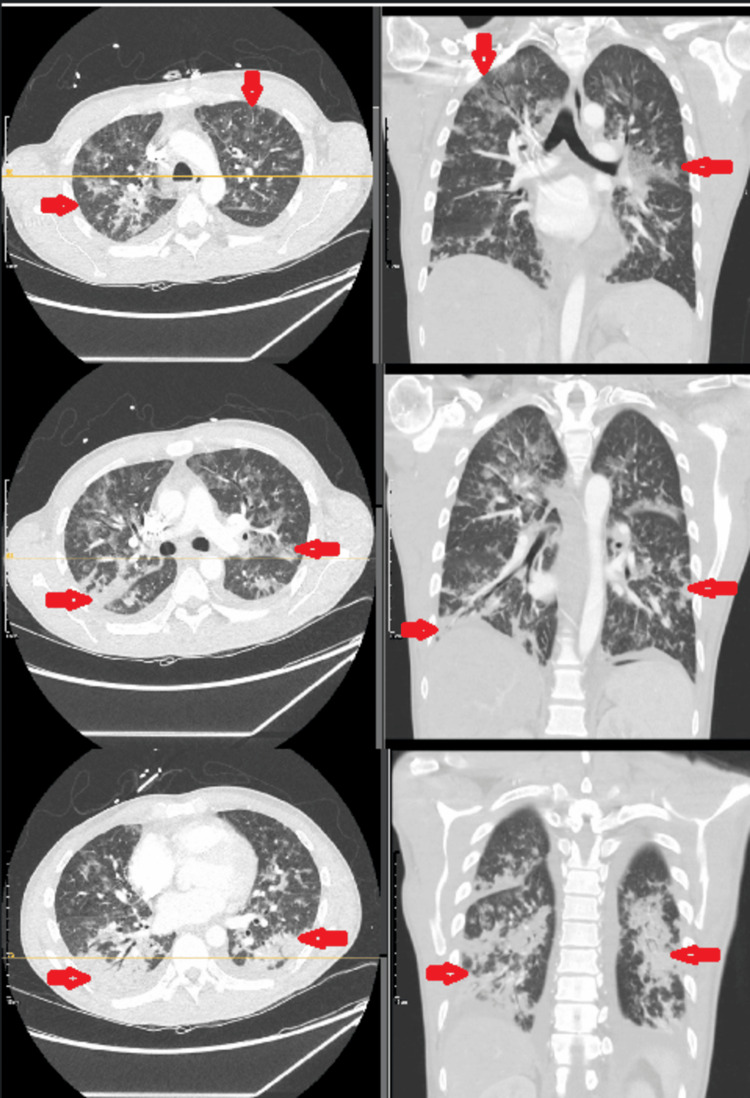 Figure 1
Figure 1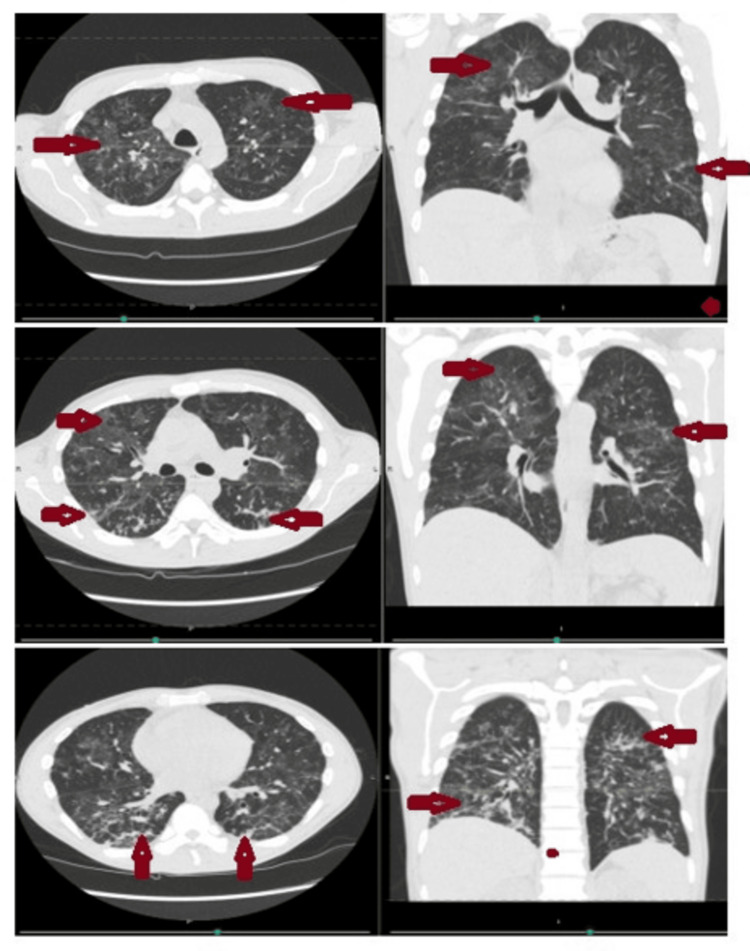 Figure 2
Figure 2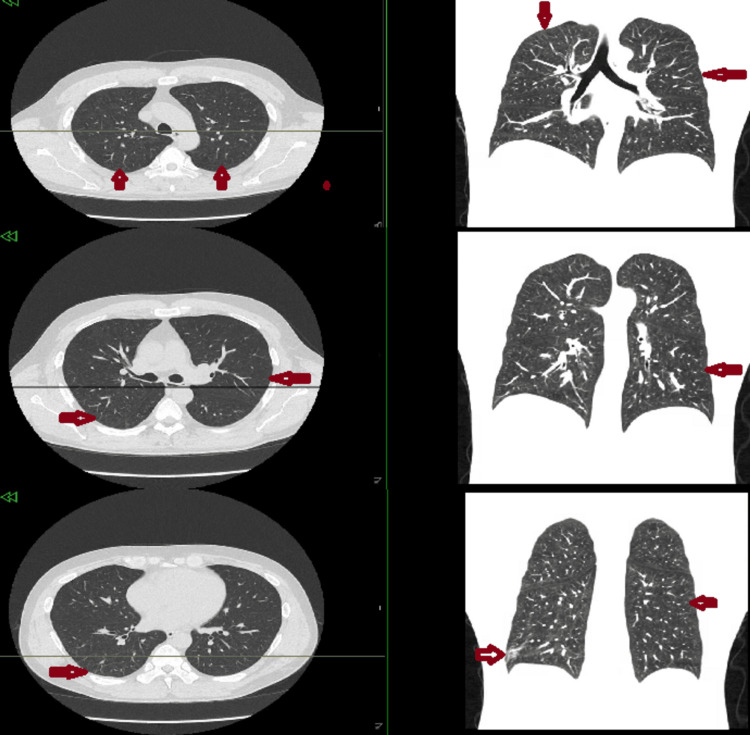 Figure 3
Figure 3| Vital sign | Patient value | Normal range |
| Temperature | Afebrile | 36.5-37.5 °C (97.7-99.5 °F) |
| Blood pressure | 110s/60s | ~120/80 mmHg |
| Heart rate | 130 beats per minute | 60-100 beats per minute |
| Oxygen saturation | 85 % on 2 L nasal cannula | ≥94 % on room air |
| Test | Patient value | Flag | Reference range |
| Hemoglobin | 6.8 g/dL | Low | 13.5-17.5 g/dL (male) |
| Hematocrit | 21.5 % | Low | 41-53 % (male) |
| Mean corpuscular volume | 105.7 fL | High | 80-100 fL |
| Red cell distribution width | 22.0 % | High | 11.5-14.5 % |
| White blood cell count | 10.0 K/µL | Normal | 4.0-11.0 K/µL |
| Platelets | 284 K/µL | Normal | 150-450 K/µL |
| Sodium | 129 mEq/L | Low | 135-145 mEq/L |
| Potassium | 5.0 mEq/L | High | 3.5-5.0 mEq/L |
| Chloride | 103 mEq/L | Normal | 98-107 mEq/L |
| Creatinine | 0.44 mg/dL | Low | 0.7-1.3 mg/dL |
| Estimated glomerular filtration rate | 137 mL/min | Normal | >60 mL/min |
| Calcium | 9.4 mg/dL | Normal | 8.5-10.5 mg/dL |
| Albumin | 3.4 g/dL | Low | 3.5-5.0 g/dL |
| Aspartate aminotransferase | 273 U/L | High | 10-40 U/L |
| Alanine aminotransferase | 83 U/L | High | 7-56 U/L |
| Alkaline phosphatase | 44 U/L | Normal | 44-147 U/L |
| Bilirubin total | 0.30 mg/dL | Normal | 0.1-1.2 mg/dL |
| Magnesium | 1.8 mg/dL | Normal | 1.7-2.2 mg/dL |
| T3 free | 2.00 pg/mL | Low | 2.1-4.2 pg/mL |
| T4 free | 1.20 ng/dL | Normal | 0.8-2.0 ng/dL |
| Thyroid-stimulating hormone | 2.476 µIU/mL | Normal | 0.3-5.0 µIU/mL |
| D-dimer | 1744 ng/mL | High | <500 ng/mL |
| Urinalysis protein | 10 mg/dL | Abnormal | Negative |
| Urinalysis nitrite | Negative | Normal | Negative |
| Urinalysis leukocyte esterase | Negative | Normal | Negative |
| Respiratory viral PCR panel | Not detected | Normal | Negative |
| Bacterial PCR panel | Not detected | Normal | Negative |
| Test | Result | Flag | Reference range |
| Procalcitonin | 0.350 ng/mL | Elevated | <0.05 ng/mL |
| C-reactive protein | 42 mg/L | Elevated | <5 mg/L |
| Erythrocyte sedimentation rate | 48 mm/hr | Elevated | <20 mm/hr (men) |
| Ferritin | 1072.5 ng/mL | High | 30-400 ng/mL |
| Test | Patient value | Flag | Reference range |
| Antinuclear antibodies screen | Positive | High | Negative |
| Anti-dsDNA antibody qualitative | Positive | High | Negative |
| Anti-dsDNA antibody quantitative | 21 IU/mL | High | <10 IU/mL |
| Anti-RNP antibody qualitative | Positive | High | Negative |
| Anti-RNP antibody quantitative | 3.4 AI | High | <0.9 AI |
| Anti-Sm/RNP antibody qualitative | Positive | High | Negative |
| Anti-Sm/RNP antibody quantitative | 7.6 AI | High | <0.9 AI |
| Anti-chromatin antibody qualitative | Positive | High | Negative |
| Anti-chromatin antibody quantitative | 1.7 AI | High | <0.9 AI |
| Anti-centromere B antibody qualitative | Negative | - | Negative |
| Anti-centromere B antibody quantitative | <0.2 AI | - | <0.9 AI |
| Anti-Jo1 antibody qualitative | Negative | - | Negative |
| Anti-Jo1 antibody quantitative | <0.2 AI | - | <0.9 AI |
| Anti-ribosomal P antibody qualitative | Negative | - | Negative |
| Anti-ribosomal P antibody quantitative | 0.2 AI | - | <0.9 AI |
| Anti-Scl-70 antibody qualitative | Negative | - | Negative |
| Anti-Scl-70 antibody quantitative | <0.2 AI | - | <0.9 AI |
| Anti-Smith antibody qualitative | Negative | - | Negative |
| Anti-Smith antibody quantitative | 0.4 AI | - | <0.9 AI |
| Anti-SSA/Ro antibody qualitative | Negative | - | Negative |
| Anti-SSA/Ro antibody quantitative | 0.4 AI | - | <0.9 AI |
| Anti-SSB/La antibody qualitative | Negative | - | Negative |
| Anti-SSB/La antibody quantitative | <0.2 AI | - | <0.9 AI |
| Rheumatoid factor | 182.1 IU/mL | High | <14 IU/mL |
| Cyclic citrullinated peptide IgG antibody | 16 units | Normal | Negative <20 units; weak positive 20-39; moderate positive 40-59; strong positive ≥60 |
| HIV 1/2 antibody/antigen screen | Nonreactive | Normal | Nonreactive |
| Complement component 3 | 74 mg/dL | Low | 90-180 mg/dL |
| Complement component 4 | 16-27 mg/dL | Normal | 10-40 mg/dL |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsInterstitial Lung Diseases and Idiopathic Pulmonary Fibrosis · Inflammatory Myopathies and Dermatomyositis · Systemic Sclerosis and Related Diseases
Introduction
Mixed connective tissue disease (MCTD), an autoimmune overlap condition characterized by high-titer anti-U1 ribonucleoprotein (U1 RNP) antibodies and clinical features resembling systemic lupus erythematosus, systemic sclerosis, and polymyositis, often presents with significant pulmonary involvement [1]. Interstitial lung disease (ILD) occurs in nearly half of patients and represents the leading cause of morbidity and mortality in MCTD [2]. Among the ILD subtypes, nonspecific interstitial pneumonia (NSIP) is the most frequently encountered pattern and is associated with ground-glass opacities, reticulation, and variable fibrosis on imaging [3].
Early recognition of NSIP is essential because timely immunosuppressive therapy can reverse inflammation and prevent irreversible fibrosis. However, NSIP often mimics infectious pneumonia, leading to diagnostic delays, inappropriate antimicrobial therapy, and progression of the underlying autoimmune disease [4,5]. Diagnostic evaluation requires integration of serologic markers, high-resolution CT (HRCT), and bronchoalveolar lavage (BAL) to exclude infection and histopathologic confirmation when necessary [6].
This case highlights an acute NSIP presentation as the first manifestation of MCTD, emphasizing the importance of early autoimmune evaluation in patients with diffuse lung opacities and persistent hypoxemia despite appropriate antibiotic therapy.
Case presentation
The patient is a 42-year-old man with a five-pack-year smoking history, pulmonary nodules, deep vein thrombosis (DVT), chronic anemia, an alcohol use disorder, and a possible diagnosis of rheumatoid arthritis (RA). He presented to the emergency department after experiencing a two- to three-day history of worsening cough, dyspnea, and generalized weakness. Additionally, he reported an unintentional weight loss of 20 pounds over the past three months.
On arrival, the patient was afebrile. Blood pressure was slightly lower than expected for a healthy adult, and his heart rate was markedly elevated, consistent with significant tachycardia. Oxygen saturation showed significant hypoxemia despite supplemental oxygen via nasal cannula (Table 1). Physical examination revealed tachypnea, tachycardia, and bilateral crackles on lung auscultation.
Initial laboratory evaluation demonstrated severe anemia, hyponatremia, and marked elevation of liver enzymes consistent with transaminitis (Table 2). The anemia showed a macrocytic pattern with significant variability in red blood cell morphology, suggesting contributions from nutritional deficiency, chronic disease, or alcohol-related marrow suppression. Hyponatremia was consistent with systemic inflammation, reduced oral intake, or possible endocrine dysfunction. Liver enzyme elevation demonstrated a pattern often seen with alcohol-associated injury, systemic inflammation, or muscle involvement related to connective tissue disease. The D-dimer was elevated, which can reflect active systemic inflammation, impaired clearance in the setting of liver dysfunction, or underlying thrombotic risk in a patient with a known history of DVT.
Thyroid function testing was performed to assess for hypothyroidism as a potential contributor to the hyponatremia. Urinalysis demonstrated the presence of protein, which, although mild, may reflect systemic inflammation, early glomerular involvement related to autoimmune disease, or transient functional proteinuria associated with acute illness. In the context of connective tissue disease, low-level proteinuria can also signal early immune-mediated renal involvement and therefore warrants continued outpatient monitoring.
A chest CT angiography scan ruled out pulmonary embolism but demonstrated extensive bilateral consolidation, ground-glass opacities, and nodular infiltrates, raising concern for ILD (Figure 1).
Axial and coronal views of chest CT angiography at three different levels, showing extensive bilateral consolidation, ground-glass, and nodular opacities (marked by red arrows)
The patient received one unit of packed red blood cells and was empirically started on intravenous cefepime and doxycycline. He was initially admitted to the medical floor but was transferred to the intensive care unit the following morning due to increasing work of breathing, tachypnea, and worsening hypoxemia. Family history was notable for dermatomyositis in a first-degree relative, suggesting a genetic predisposition.
Bronchoscopy with transbronchial biopsies under fluoroscopic guidance was performed in the posterior segment of the right upper lobe, the lateral segment of the right middle lobe, and the lateral basal segment of the right lower lobe. Biopsy results revealed uniform interstitial inflammation and fibrosis consistent with NSIP. BAL was negative for bacterial, viral, and fungal pathogens, supporting a noninfectious inflammatory etiology. Laboratory evaluation showed elevated inflammatory markers, including procalcitonin, C-reactive protein, erythrocyte sedimentation rate, and ferritin levels (Table 3).
Rheumatology was consulted after an autoimmune serologic and rheumatologic workup, which returned positive for antinuclear antibody (ANA), anti-double-stranded DNA, anti-ribonucleoprotein (anti-RNP), anti-Smith/ribonucleoprotein (anti-Sm/RNP), anti-chromatin antibodies, and rheumatoid factor, with a negative cyclic citrullinated peptide IgG antibody (anti-CCP) (Table 4). This autoantibody profile, particularly the presence of anti-RNP and anti-Sm/RNP together with high-titer ANA, supports a possible diagnosis of MCTD, which is classically associated with antibodies targeting U1-RNP and overlapping features of several connective tissue diseases.
Table 4: Laboratory investigations: autoimmune serology and rheumatologic workupAI stands for Antibody Index. It is a standardized unit used by many autoimmune serology laboratories to report the quantitative level of an antibody: (1) AI < 0.9 → Negative, (2) AI 1.0-1.5 → Weak positive, and (3) AI > 1.5 → Strong positive. Different laboratories may have slightly different cutoffs, but the concept is the same.ANA, antinuclear antibody; anti-dsDNA, anti-double-stranded DNA; anti-RNP, anti-ribonucleoprotein; anti-Sm/RNP, anti-Smith/ribonucleoprotein
In this context, the positive RF with a negative anti-CCP may reflect involvement of the MCTD spectrum rather than classic seropositive RA, because anti-CCP antibodies are much more specific for RA and are detected in only a minority of patients with MCTD. The patient had a similar serologic pattern of positive RF with negative anti-CCP several years earlier; at that time, additional autoimmune testing was not performed, and he declined medical management with methotrexate or hydroxychloroquine.
Given the clinical presentation, radiographic findings, biopsy results, and autoimmune serologies, and to address his acute symptoms, a treatment regimen was initiated beginning with intravenous methylprednisolone sodium succinate at a dosage of 60 mg every eight hours for two days. This was followed by a higher dosage of 500 mg daily for three days based on pulmonary recommendations. All cultures, including BAL samples tested for acid-fast bacilli, remained negative, supporting continued immunosuppressive treatment. The rheumatology team was also involved in the patient’s care and concurred with the management plan.
The patient’s respiratory status, including work of breathing and hypoxemia, continued to improve with corticosteroids, and he was weaned off supplemental oxygen. He was transitioned to oral prednisone at a dose of 60 mg daily with a tapering schedule. Mycophenolate mofetil (MMF) was initiated after confirming appropriate baseline studies, including a complete blood count, renal function panel, and hepatic function panel. MMF was started at 500 mg orally twice daily and gradually increased to a therapeutic dose of 1,000 mg twice daily for long-term management of suspected MCTD-associated NSIP. This treatment approach aligns with current recommendations from the American College of Rheumatology and the American College of Chest Physicians.
Transthoracic echocardiography was performed to rule out a cardiac cause of tachycardia. It revealed sinus tachycardia as fast as 120 beats per minute during the study, with normal overall and regional left ventricular systolic function, no valvular disease, no right heart disease, and no signs of pulmonary hypertension. Sinus tachycardia was considered secondary to anemia, anxiety, and acute illness and resolved as the patient improved.
He was discharged with close outpatient follow-up arranged with pulmonology, rheumatology, and gastroenterology for continued monitoring of his ILD and autoimmune disease and was scheduled for a colonoscopy as an outpatient to evaluate chronic anemia. Additionally, he reported bilateral lower extremity paresthesias suggestive of autoimmune-related neuropathy, for which gabapentin was initiated. Outpatient electromyography and neurology follow-up were recommended.
A one-month follow-up chest CT showed marked improvement in the multifocal consolidations, ground-glass changes, and nodular opacities (Figure 2), with near-complete resolution demonstrated on the six-month follow-up CT scan (Figure 3).
Axial and coronal views of chest CT scan without contrast at three different levels, showing improvement in bilateral consolidation, ground-glass, and nodular opacities, as indicated by the red arrows
Axial and coronal views of chest CT scan without contrast at three different levels, highlighting interval resolutionRed arrows mark the same previously involved areas for comparison.
Discussion
NSIP is the most prevalent ILD pattern observed in MCTD and may occur in up to two-thirds of affected patients [2]. NSIP is characterized radiographically by ground-glass opacities, reticulations, and traction bronchiectasis, and histologically by temporally uniform inflammation with minimal architectural distortion [3]. These features differentiate NSIP from usual interstitial pneumonia (UIP), which is associated with worse outcomes and limited responsiveness to immunosuppression [4].
The development of ILD in MCTD is attributed to immune-mediated epithelial injury, chronic antigenic stimulation, microvascular dysfunction, and reflux-related microaspiration. Autoantibodies such as anti-U1 RNP, anti-Ro52, and anti-Sm are associated with a higher risk and greater severity of lung involvement [5,6]. Our patient’s serologic findings and family history align with previously described risk factors.
A major diagnostic challenge lies in distinguishing NSIP from infectious pneumonia. Both conditions may present with bilateral ground-glass opacities and respiratory failure; however, NSIP typically lacks significant leukocytosis, persistent fever, or microbiologic evidence of infection. The American Thoracic Society and European Respiratory Society (ERS) guidelines emphasize the importance of bronchoscopy with BAL to exclude infection before initiating high-dose steroids [7].
HRCT plays a pivotal role in NSIP diagnosis. Findings such as diffuse ground-glass opacities without honeycombing suggest an inflammatory phenotype that responds well to immunosuppression [3,8]. Furthermore, the updated ERS ILD nomenclature underscores the importance of identifying fibrosing versus inflammatory patterns early to guide long-term management [9].
Corticosteroids remain the first-line treatment for inflammatory NSIP. Steroid-sparing agents such as MMF or azathioprine are commonly used for long-term maintenance, supported by data demonstrating improved stabilization of lung function and reduced relapse [10,11]. Patients with rapidly progressive or refractory ILD may benefit from cyclophosphamide or rituximab [12]. Our patient’s rapid improvement following corticosteroids and mycophenolate is consistent with favorable outcomes reported for inflammatory NSIP in connective tissue disease-associated ILD (CTD-ILD) cohorts.
Prognosis varies by the extent of fibrosis and response to therapy. NSIP generally carries a more favorable prognosis than UIP, especially when treated early [2,13]. Ongoing surveillance with pulmonary function testing and HRCT is essential given the potential for relapse or fibrotic progression [14]. This case reinforces the importance of considering autoimmune ILD in patients with diffuse opacities and unexplained hypoxemia, particularly when they fail to respond to antibiotics [15].
Conclusions
This case emphasizes the importance of considering CTD-ILD in patients presenting with diffuse ground-glass opacities, atypical respiratory symptoms, or failure to improve with appropriate antimicrobial therapy. NSIP is the most common ILD subtype in MCTD and may present acutely with severe hypoxemia. Early identification of autoimmune features, comprehensive serologic testing, and exclusion of infectious etiologies are essential steps in the diagnostic approach.
Prompt initiation of corticosteroids, along with appropriate steroid-sparing agents such as MMF or azathioprine, can lead to rapid clinical improvement, radiologic resolution, and prevention of irreversible fibrosis. Multidisciplinary collaboration among pulmonology, rheumatology, radiology, and pathology is crucial to optimize outcomes. Clinicians should maintain a high index of suspicion for CTD-ILD in cases of nonresolving pneumonia or unexplained hypoxemic respiratory failure, as early recognition and treatment can substantially alter disease trajectory and long-term prognosis.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Interstitial lung disease in mixed connective tissue disease: an advanced search Cureus Santacruz JC Mantilla MJ Rodriguez-Salas G Rueda I Pulido S Varela DC Londono J 015202310.7759/cureus.36204 PMC 1010381037065288 · doi ↗ · pubmed ↗
- 2The other connective tissue disease-associated interstitial lung diseases: Sjogren's syndrome, mixed connective tissue disease, and systemic lupus erythematosus Curr Opin Pulm Med Lee CT Strek ME 3883952720213412762010.1097/MCP.0000000000000791 PMC 8373683 · doi ↗ · pubmed ↗
- 3Interstitial lung disease associated with connective tissue diseases Lung Inflammation in Health and Disease, Volume II. Advances in Experimental Medicine and Biology Peredo RA Mehta V Beegle S Cham Springer 1304202110.1007/978-3-030-68748-9_534019264 · doi ↗ · pubmed ↗
- 4Diagnosis and treatment of connective tissue disease-associated interstitial lung disease Chest Vij R Strek ME 81482414320132346015910.1378/chest.12-0741 PMC 3590889 · doi ↗ · pubmed ↗
- 5Characterization of interstitial lung disease associated with anti-ribonucleoprotein antibodies J Clin Rheumatol Lhote R Grenier P Haroche J 3273332620203141547610.1097/RHU.0000000000001127 · doi ↗ · pubmed ↗
- 6Interstitial lung disease in patients with mixed connective tissue disease: pilot study on predictors of lung involvement Clin Exp Rheumatol Narula N Narula T Mira-Avendano I Wang B Abril A 648651362018 https://pubmed.ncbi.nlm.nih.gov/29745875/29745875 · pubmed ↗
- 7Diagnosis of idiopathic pulmonary fibrosis. An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline Am J Respir Crit Care Med Raghu G Remy-Jardin M Myers JL 068198201810.1164/rccm.201807-1255 ST 30168753 · doi ↗ · pubmed ↗
- 8Interstitial lung disease in connective tissue disorders Lancet Fischer A du Bois R 68969838020122290189010.1016/S 0140-6736(12)61079-4 · doi ↗ · pubmed ↗