Identification of a culturable fungal species and endosymbiotic bacteria in saliva of Aedes aegypti and Culex pipiens and their impact on arbovirus infection in vitro
Lanjiao Wang, Laure Remue, Nikki Adriaens, Alina Soto, Sam Verwimp, Joyce van Bree, Katrien Trappeniers, Leen Delang

TL;DR
This study identifies fungi and bacteria in mosquito saliva and shows that these microbes can influence arbovirus infection in human cells.
Contribution
The study is the first to identify culturable fungi and endosymbiotic bacteria in the saliva of Aedes aegypti and Culex pipiens mosquitoes.
Findings
Penicillium crustosum was identified as a fungal species in mosquito saliva.
Saliva bacterial communities were more diverse than those in the midgut.
Antibiotic or antifungal treatment reduced microbes in saliva and decreased viral infection in human cells.
Abstract
Mosquito saliva plays a key role in arbovirus transmission and pathogenesis. It was shown that saliva contains several molecules that are essential for blood feeding. Recently, bacteria were also reported to be present in the saliva of Aedes albopictus and Anopheles mosquitoes. Nevertheless, information on the bacterial communities in Aedes and Culex saliva is still scarce. This study isolated and identified culturable fungal and bacterial colonies from saliva harvested from Aedes aegypti (laboratory strain) and Culex pipiens (field-collected) mosquitoes. 16S metagenomic sequencing was performed to identify bacterial communities in saliva and mosquito organs. Furthermore, it was assessed how these microbial communities were affected upon blood feeding and upon oral treatment with antibiotics and an antifungal drug. The fungal species Penicillium crustosum was identified in mosquito…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
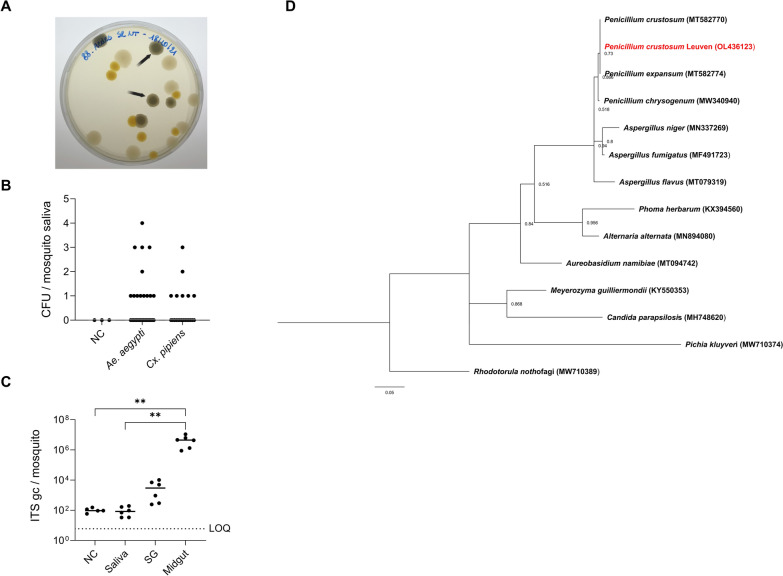 Figure 1
Figure 1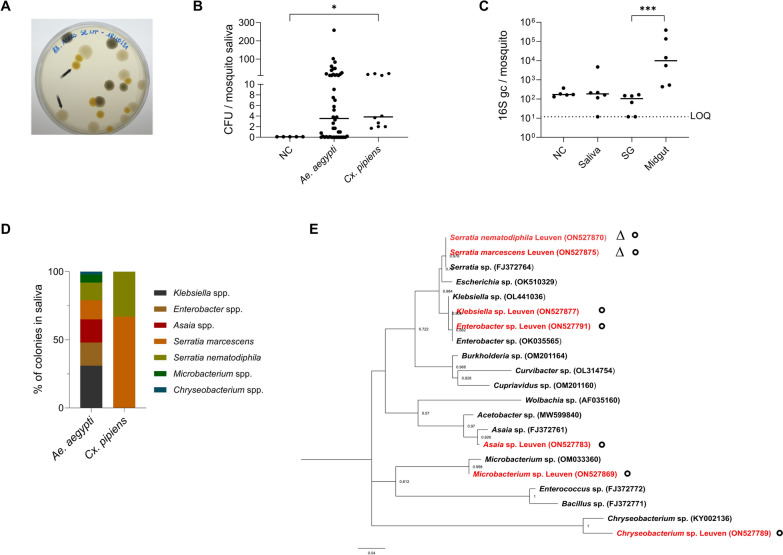 Figure 2
Figure 2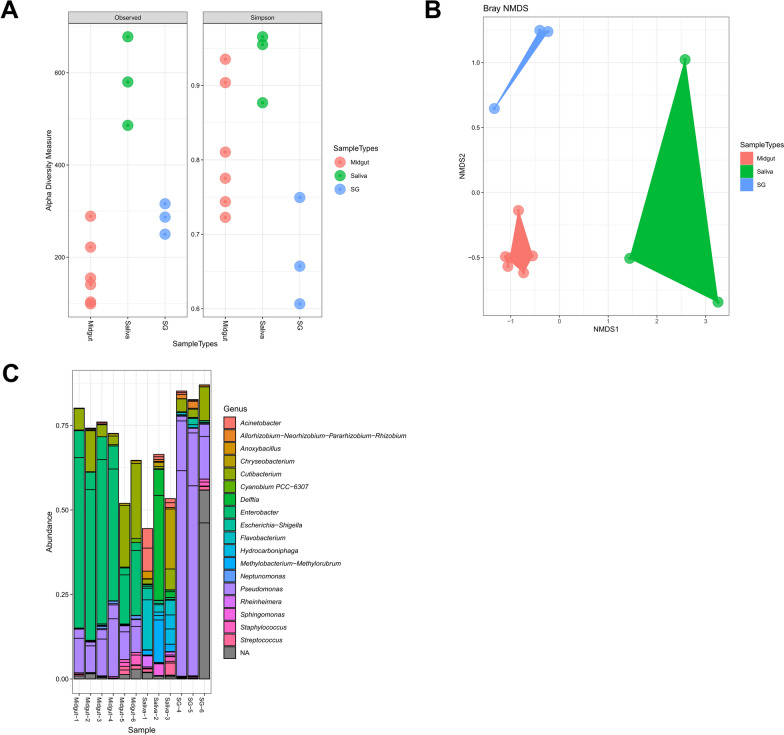 Figure 3
Figure 3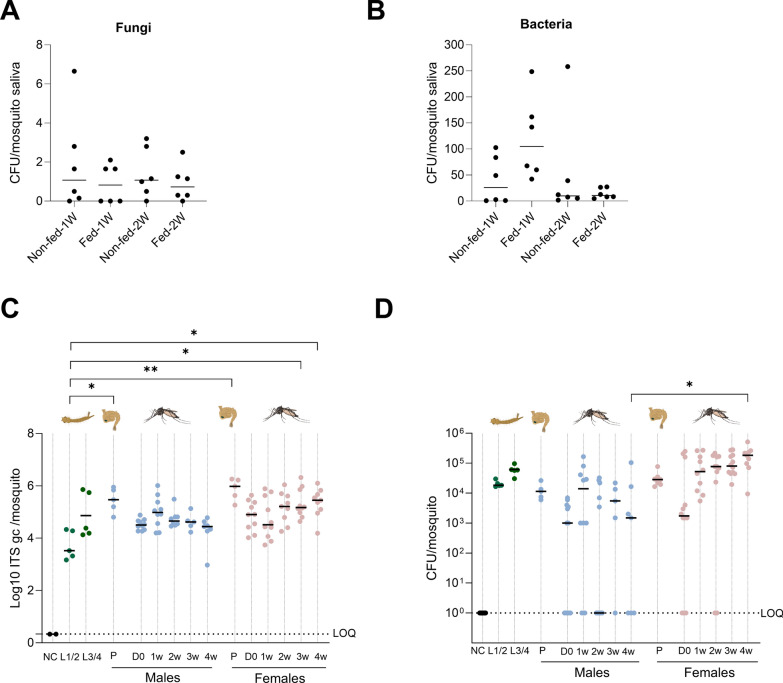 Figure 4
Figure 4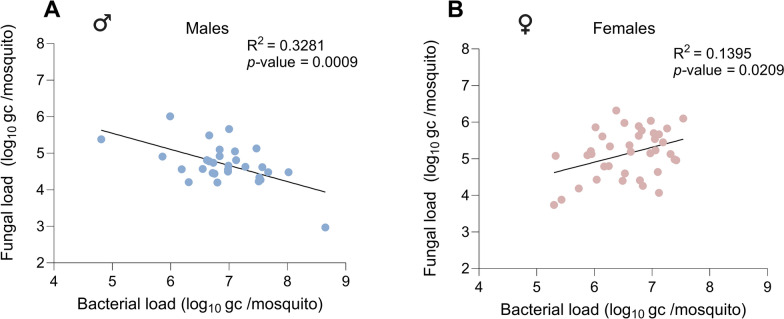 Figure 5
Figure 5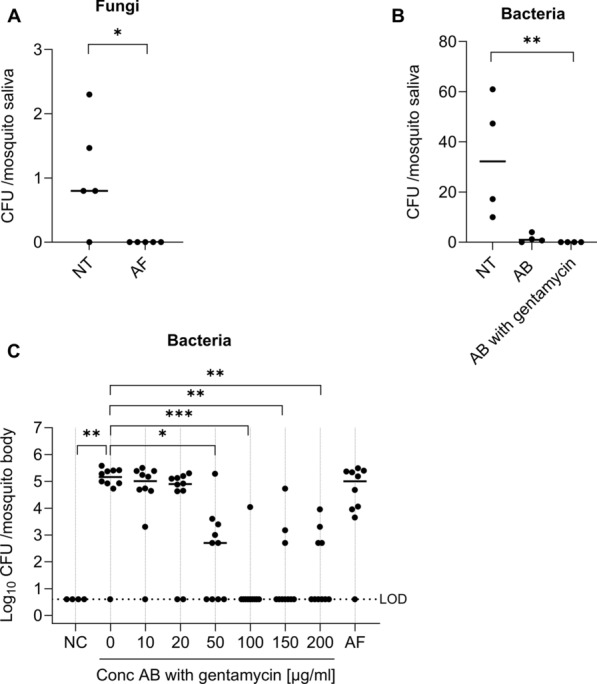 Figure 6
Figure 6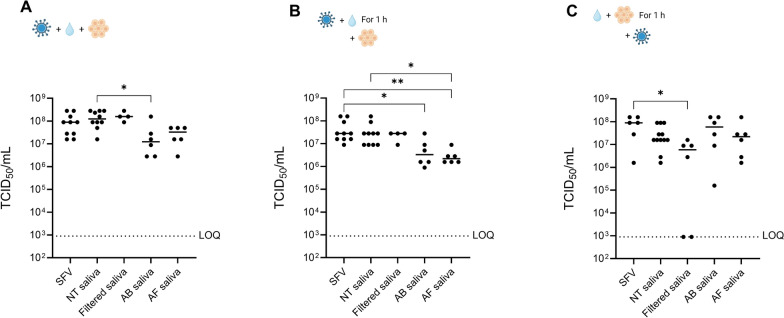 Figure 7
Figure 7 Figure 8
Figure 8- —https://doi.org/10.13039/501100004040KU Leuven
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsInsect symbiosis and bacterial influences · Entomopathogenic Microorganisms in Pest Control · Vector-borne infectious diseases
Background
Mosquitoes of the Culicidae family are considered the deadliest animals on the planet [1], owing to the ability of female mosquitoes to transmit pathogens such as arboviruses (i.e., arthropod-borne viruses) during blood feeding. The primary mosquito vectors of medically important arboviruses include Aedes aegypti, which transmits chikungunya (CHIKV), Zika (ZIKV), dengue (DENV), and yellow fever (YFV) viruses, and Culex pipiens, which is the principal vector of West Nile virus (WNV) [1]. Arboviruses are an emerging threat to the health of humans and animals worldwide [2]. Symptoms caused by these viruses can vary from asymptomatic or mild febrile illness to severe diseases, such as chronic arthralgia and arthritis (CHIKV), congenital microcephaly and Guillain–Barré syndrome (ZIKV), dengue hemorrhagic fever and shock syndrome (DENV), and viral encephalitis (WNV), which can potentially be fatal [3–5]. In recent decades, arbovirus epidemics have increased as a consequence of the development of global transportation systems, deforestation, urbanization, and an increasing human population density.
Several decades of global efforts toward the development of arbovirus vaccines have resulted in few successful candidates. Furthermore, there are currently no approved, specific antiviral drugs available to treat arbovirus infections. Therefore, the current primary strategy to control arbovirus outbreaks in high-risk countries relies on mosquito vector control with insecticides aiming at reducing contact with potentially infected mosquitoes [6]. Owing to the long-term usage of insecticides of the same chemical classes, including organophosphates and synthetic pyrethroids, insecticide-resistant mosquitoes are widely spread and are considered a major threat to vector control by the World Health Organization [7]. Other known limitations of insecticide use include suboptimal timing of application, limited efficacy, low community acceptance, and high economic costs, as well as potential off-target effects on nonvector insect species [8]. Therefore, new strategies for vector control are needed to reduce the global burden caused by arboviruses and other vector-borne pathogens. Promising results have been accomplished using host-associated microorganisms (i.e., microbiota members) as novel vector control strategies [9, 10].
When feeding on an infected host, the female mosquito ingests an arbovirus simultaneously with the blood into the midgut. To be efficiently transmitted to another vertebrate host, the virus must replicate primarily in the midgut and then successfully disseminate to secondary organs, especially to the salivary glands. The virus must cross both the midgut and salivary gland barriers to reach and replicate in salivary glands. Finally, to infect a new host, the virus needs to be released into the mosquito saliva and deposited into the skin during a subsequent blood feeding [11]. Hence, the transmission of viruses through mosquitoes among diverse hosts can be influenced not only by extrinsic factors such as temperature, humidity, and nutrition but also by intrinsic mosquito-related factors including innate immune responses, microbiota, and genetic factors [12].
The midgut and the salivary glands have been characterized as key barriers to systemic arbovirus infection in the mosquito and to transmission within saliva. Meanwhile, both organs are also known to be colonized by dynamic microbial communities [13–16]. Most studies have focused on how gut microbiota impact mosquito biology, including vector competence [17]. Multiple interactions between microbial species and arboviruses have been demonstrated. For example, Serratia odorifera, a bacterium inhabiting the midgut of Ae. aegypti, has been shown to enhance mosquito susceptibility to DENV and CHIKV by suppressing the mosquito immune response [18, 19]. Furthermore, Serratia marcescens was identified as a midgut commensal bacterium with the ability to promote mosquito permissiveness to arboviruses by secreting SmEnhancin, a protein to digest the mosquito midgut membrane-bound mucins [20]. In addition to bacteria, a Talaromyces fungus was found in the midgut of field-captured Ae. aegypti, rendering the mosquito more permissive to DENV infection by downregulating digestive enzyme genes and their activity in the midgut [21]. Although the gut forms a first important barrier in establishing a viral infection in the mosquito, viral transmission to the next mammalian host occurs via the mosquito saliva. While biting a host, female mosquitoes probe into the host skin in search of a blood vessel and meanwhile inject saliva into the epidermis and dermis of the vertebrate host. Mosquito saliva contains vasodilatory, anti-hemostatic, angiogenic, and anti-inflammatory molecules that ensure continuous blood pumping to the mosquito during feeding [22, 23]. In addition, mosquito saliva contains molecules that could alert the immune system of the vertebrate host and potentially block viral transmission. Therefore, mosquito saliva components are being studied as promising candidates for vaccine development [24]. Interestingly, two recent papers demonstrated that the saliva of Aedes albopictus and Anopheles mosquitoes contains bacteria [25, 26]. Nevertheless, our current understanding of which microbiota members are present in the saliva of Aedes and Culex mosquitoes is still very limited. Furthermore, it is unclear whether and how the saliva microbiota impacts vector competence and—being transferred to the mammalian host—if, downstream, they affect the virus replication and virus infection in the host.
Here, we describe that both fungi and bacteria can be cultured from the saliva of laboratory-maintained Ae. aegypti and field-collected Cx. pipiens mosquitoes. In addition, oral treatment of adult mosquitoes with antibiotics or an antifungal drug resulted in a significant reduction of bacteria or fungi in saliva. Finally, we demonstrated that saliva from antibiotic- or antifungal-treated mosquitoes decreased infectious viral loads of Semliki Forest virus (SFV) in human skin fibroblasts. Further in vivo studies are required to better understand its impact on viral infection.
Methods
Mosquito strains
The Ae. aegypti Paea laboratory strain (provided by Pasteur Institute Paris via the EU-funded Infravec consortium) was reared and maintained under controlled conditions (28 ± 1 °C, relative humidity of 80%, light:dark cycle of 16 h:8 h). Field Cx. pipiens mosquitoes were collected in Leuven (N 50°52′41, E 4°41′21, Belgium) during the summer of 2021. Adult mosquitoes were trapped with BG-Sentinel traps (BioGents GmbH, Germany) with the help of BG-lure (BioGents GmbH, Germany). After sorting mosquitoes on the basis of morphology, Cx. pipiens was maintained under conditions of 25 ± 1 °C, relative humidity of 50%, and a light:dark cycle of 14 h:10 h. Adult mosquitoes were housed in Bugdorm-1 insect cages (30 × 30 × 30 cm, Bugdorm, Taiwan, China) and fed with 10% sterile sugar solution.
Species identification of field mosquitoes by multiplex quantitative polymerase chain reaction
Species identification was performed as described earlier [27]. In brief, wings and legs of individual female mosquitoes were homogenized by bead disruption (2.8 mm Precellys, Bertin Instruments). Subsequently, the supernatant was hydrolyzed at 95 °C for 10 min. Multiplex quantitative polymerase chain reaction (qPCR) was performed using a forward primer (5′-GCGGCCAAATATTGAGACTT-′3) and reverse primer (5′-CGTCCTCAAACATCCAGACA-′3) targeting the gene locus for acetylcholinesterase 2 to identify the Cx. pipiens complex [28]. In addition, subspecies-specific probes were used by targeting the CQ11 microsatellite locus to distinguish between Cx. pipiens pipiens (5′-GCTTCGGTGAAGGTTTGTGT-′3) [28] and Cx. pipiens molestus (5′-TGAACCCTCCAGTAAGGTATCAACTAC-′3) [29]. Reactions were prepared in a final volume of 25 µL containing 3 µL of template DNA, 6.25 µL of 2 × TaqMan^™^ PCR Master mix, 0.38 µL of each primer at 10 µM, 1 µL of each probe at 5 µM, 0.06 µL reverse transcriptase, and 12.94 µL of nuclease-free water. The thermal profile consisted of an initial step of 15 min at 95 °C, followed by 40 amplification cycles of 1 min at 94 °C, 1 min at 48 °C, and 1 min at 72 °C.
Forced salivation and dissection of midguts and salivary glands
Mosquitoes were starved 24 h prior to salivation. Female Ae. aegypti or Cx. pipiens mosquitoes were anesthetized by placing them under cold conditions (4–8 °C) for 5 min, or by CO_2_ released by dry ice for 2 min, respectively. While sedated, legs and wings were removed to immobilize the mosquito during salivation. Saliva for culturing was harvested by gently placing the proboscis of a living female into a 20-µL pipette tip (uTIP, Biotix) containing 20 µL of a 1:1 solution of 10% sterile sugar solution and 1× phosphate-buffered saline (PBS). Saliva for the in vitro infection assays was harvested by gently placing the proboscis of a living female into the pipette tip containing 1 µL silicon oil as described previously [30]. Blank tips containing only the salivation solution were placed aside on the bench as the environment control during each salivation. After 1 h of forced salivation, saliva was harvested and stored at −80 °C either individually or pooled for further studies.
The surface of the mosquito body was disinfected individually by 75% ethanol and rinsed twice with 1× PBS buffer before dissection of the salivary glands and midgut. Salivary glands were harvested before the midgut to avoid potential contamination as the midgut harbors the highest bacterial load. The salivary glands and midguts were pooled (8–12 females) in homogenization tubes (2.8 mm Precellys, Bertin Instruments) containing 150–200 µL 1× PBS and then stored at −80 °C for further analysis.
Artificial blood feeding, antibiotic, and antifungal treatment
To study the effects of a blood meal on saliva microbiota, 5–7-day-old Ae. aegypti female mosquitoes were orally fed for 30 min using an artificial membrane feeding system (Hemotek, UK). The blood meal contained fresh rabbit erythrocytes washed with PBS, and plasma was replaced by the same volume of sterile FBS. Fully engorged females were cold-anesthetized and separated to be maintained up to 2 weeks until forced salivation, under the same controlled conditions as mentioned above. In parallel, the blood meal was cultured on Luria–Bertani (LB) plates to determine the absence of bacteria in the blood meal.
Adult mosquitoes were treated with an antibiotic cocktail with doses ranging from 10 units/mL penicillin and 10 µg/mL streptomycin/gentamycin (Penicillin–Streptomycin 100X, Gentamycin, Gibco^™^) up to 200 units/mL penicillin and 200 µg/mL streptomycin/gentamycin into the 10% sugar solution for 6 days. For the antifungal treatment, 25 µg/mL of Fungin^™^ (InvivoGen) was added into the 10% sugar solution for 10 days. The sugar solution containing antibiotic and/or antifungal compounds was refreshed every day throughout the treatment.
Bacteria and fungi culturing on agar plates
Mosquito-derived samples (saliva, salivary glands, and midguts) were cultured on Luria-Bertani (LB) broth, Brain Heart Infusion (BHI) broth, or blood agar plates at 28 °C to compare microbial growth on different media. Saliva from 8–12 mosquitoes was pooled. Approximately 50 µL of the saliva pool was inoculated per agar plate. For the salivary gland samples, 50 µL of supernatant post homogenization was used. For the midgut, the supernatant was diluted 10–100 times in sterile water post homogenization before culturing 50 µL of supernatant on the agar plates. As a negative control, the salivation solution was cultured in parallel. Plates were checked after 24, 48, and 120 h of incubation for bacteria and up to 1 month for fungi. Colony-forming units (CFU) were calculated to determine the bacterial and fungal loads.
Bacterial identification by 16S Sanger sequencing
Individual colonies were selected from the agar plates and resuspended in 30 µL sterile water. The bacterial suspension was heat-inactivated at 95 °C for 15 min. When necessary, the DNA concentration was diluted to 100 ng/µL. PCR amplification of 467 bp in the 16S ribosomal RNA gene was performed with the 16S forward universal primer (5′ TCC TAC GGG AGG CAG CAG T ′3) and 16S reverse universal primer (5′ GGA CTA CCA GGG TAT CTA ATC CTG TT ′3) as described previously [31]. The PCR product was confirmed by gel electrophoresis (1% agarose gel). PCR samples were purified by the Wizard SV Gel and PCR Clean system following manufacturers’ instructions. After purification, samples were sent to Macrogen Europe for Sanger sequencing. The DNA sequences obtained were trimmed using TrimAl (v. 1.3) and searched for homology to those available at the GenBank–European Molecular Biology Laboratory (EMBL) database using the Basic Local Alignment Search Tool (BLAST) program (National Center for Biotechnology Information [NCBI]).
Fungal identification by ITS3–ITS4 and CRU-specific Sanger sequencing
A portion of the fungal colony was collected into a homogenization tube with 500 µL of PBS. Homogenization was performed at two cycles of 6800 rpm for 10 s with a pause of 20 s between each cycle. After homogenization, 200 µL of the supernatant was used for DNA extraction, which was performed using the QIAamp DNA Kit 250 (51306, QIAGEN) following manufacturers’ instructions. The universal forward primer targeting the ITS3 region (5′ GCA TCG ATG AAG AAC GCA GC ′3) and universal reverse primer targeting the ITS4 region (5′ TCC TCC GCT TAT TGA TAT GC′3) were used for ITS PCR, amplifying a 350 bp PCR product as described previously [32]. For species-specific detection of Penicillium crustosum, the CRU-F forward primer (5′ TCC CAC CCG TGT TTA TTT TA ′3) and CRU-R reverse primer (5′ TCC CTT TCA ACA ATT TCA CG ′3) were used, amplifying a 892 bp PCR product [33]. The length of the PCR products was confirmed by gel electrophoresis (1% agarose gel). PCR samples were purified by the Wizard SV Gel and PCR Clean system (Promega) following manufacturers’ instructions. The protocol for Sanger sequencing and analysis is identical as described above for the bacterial identification.
DNA extraction from midgut, salivary gland, and saliva samples
DNA extractions were strictly performed under a laminar flow cabinet. Blank Eppendorf tubes were added per experiment as a negative environmental control. DNA extractions were performed using the QIAamp DNA Kit 250 (51306, QIAGEN) following an adapted version of the manufacturers’ protocol including an additional step of adding 0.5 µL of Ready-Lyse^™^ Lysozyme Solution (Lucigen) to enable the collection of DNA from both Gram-positive and Gram-negative species. Next, 100 µL ATL buffer and 20 µL proteinase K were added to each sample, followed by an incubation step at 56 °C for 5 min. After pulse-vortexing for 15 s, samples were incubated at 70 °C for 10 min and 230 µL ethanol was added. The following steps followed the original protocol according to manufacturers’ instructions. Samples were eluted in 50 µL AE buffer and were stored at −20 °C until further analysis.
Quantification of bacterial and fungal loads by qPCR
The bacterial and fungal loads in extracted DNA samples were quantified by qPCR using the iTaq Universal SYBR Green One-Step kit (Bio-Rad). Each reaction was prepared in a final volume of 20 µL containing 5 µL template DNA, 10 µL 2× one-step SYBR Green reaction mix, 0.4 µL of each primer at 10 µM, and 4.2 µL of nuclease-free water. The same primers were used as for Sanger sequencing (described above). The thermal profile consisted of an initial step of 10 min at 95 °C, followed by 40 amplification cycles of 15 s at 95 °C and 1 min at 50 °C for bacteria, while a 45 °C annealing temperature was used for fungal quantification. The genome copies/mosquito for each sample were quantified on the basis of the use of a gBlocks^™^ Gene Fragment (IDT) as a standard dilution for both the 16S and ITS qPCR. Limit of detection (LOD) was determined as the lowest bacterial/fungal load able to be detected by the qPCR assay in 95% of experiments.
Phylogenetic analysis
Sanger sequencing was performed by Macrogen Europe. The consensus sequence of fungal or bacterial colonies together with reference sequences from GenBank were aligned with MEGA-X. The resulting alignment was trimmed by using TrimAl (v.1.3). A maximum likelihood (ML) tree was constructed using MEGA-X with the substitution model being Kamura 2-parameter model (K2) and Gamma distributed (G). A total of 500 bootstrap replicates were used. Finally, trees were visualized using FigTree v1.4.4.
16S metagenomic sequencing analysis
DNA extractions were performed using the QIAamp DNA Kit 250, including an additional step of adding Ready-Lyse^™^ Lysozyme Solution as described above. Subsequently, the DNA was sent to Macrogen company to perform the following pipeline: Library prep (16S rRNA V3–V4 Illumina) and Sequencing (MiSeq—300 paired-end; 200k reads/100k paired-end reads). High-quality sequences were obtained from Macrogen. Raw data were analyzed via the DADA2 pipeline. In brief, paired-end sequences were checked, de-replicated, filtered, and merged, and chimera sequences were removed (consensus) using the DADA2 quality control method. The putative error-free sequences were referred to as sequence variants (SVs). Sequenced samples were provided as amplicon sequence variant (ASV) abundance tables for further analysis. SVs were assigned taxonomy using the assignTaxonomy function within DADA2 with the naive Bayesian classifier method, trained on the Silva version 138 99% database, into phylum, class, order, family, and genus levels to evaluate the corresponding taxonomic abundance.
Virus strains and cells
The SFV Vietnam strain (Genbank EU350586.1) belongs to the collection of the Rega Institute for Medical Research (Belgium). Virus stocks were propagated in Ae. albopictus-derived C6/36 cells (ATCC CRL-1660) at 28 °C and stored at −80 °C. Viral titers were determined by endpoint titration and plaque assay on Vero cells. African green monkey kidney epithelial cells (Vero cells, ATCC CCL-81) were maintained in minimal essential medium (MEM, Gibco) supplemented with 10% fetal bovine serum (FBS, Gibco), 1% l-glutamine (L-glu, Gibco), 1% sodium bicarbonate (NaHCO_2_, Gibco), and 1% nonessential amino acids (NEAA, Gibco). Human skin fibroblasts (ATCC CRL-2522) were grown in MEM supplemented with 10% FBS, 1% L-glu, 1% NaHCO_2_, 1% NEAA, and 1% sodium pyruvate (Gibco). Cell cultures were maintained at 37 °C in an atmosphere of 5% CO_2_ and 95–99% relative humidity. Assays were performed using a similar medium but supplemented with 2% FBS (2% assay medium).
In vitro assays of co-incubation of mosquito saliva with Semliki Forest virus
Human skin fibroblast cells were pre-seeded (12,000 cells/well, in 2% assay medium) in 96-well-plates 24 h before the infection. Saliva from nontreated Ae. aegypti, AF saliva from antifungal-treated females, AB saliva from antibiotic-treated females, and filtered saliva from nontreated Ae. aegypti using a 0.2-µm filter were assessed. Three scenarios were evaluated: (1) cells were directly inoculated with saliva and virus multiplicity of infection (MOI:0.001) for 1 h; (2) virus was pre-incubated with saliva for 1 h at 28 °C before being added to the cells for 1 h (MOI:0.001); and (3) cells were pre-incubated with saliva for 1 h followed by virus infection (MOI: 0.001) for 1 h. After virus infection for 1 h, the virus inoculum was removed, cells were washed 3× with PBS, and, subsequently, a fresh 2% assay medium was added. The plates were maintained at 37 °C with 5% of CO_2_ until 24 h post infection, when supernatant was harvested and stored at −80 °C for further analysis. The levels of infectious virus in the supernatant were determined by endpoint titrations on Vero cells. Cells were pre-seeded in 96-well-plates (25,000 cells/well) in 2% assay medium and incubated overnight (37 °C, 5% CO_2_). The next day, tenfold serial dilutions of the samples were prepared in triplicate in the plates and incubated for 3 days (37 °C, 5% CO_2_). At day 3 post infection, cells were scored microscopically for virus-induced cytopathogenic effect (CPE). The tissue culture infectious dose_50_/ml (TCID_50_/ml) was calculated using the Reed and Muench method [34]. Limit of quantification (LOQ) was determined as the lowest TCID_50_ value that could be quantified by the endpoint titration assay.
Statistical analysis
GraphPad Prism 9.4.1 and R (version 4.1.0) were used to make graphs and perform statistical analyses. In DADA2, alpha diversity in the microbial communities identified from different organs was visualized by Phyloseq and scored using the “observed” and “Shannon” diversity indexes, and Kruskal–Wallis tests were used. Beta diversity was assessed using Bray–Curtis distances. The bacterial and fungal loads were statistically analyzed using the Mann–Whitney test (two groups) or the Kruskal–Wallis test (three groups and more) followed by Dunn’s multiple comparisons with Bonferroni correction. Statistical details are described in the figure legends. The statistical significance threshold was assessed at P < 0.05.
Results
Identification of fungi in mosquito saliva
Mosquito saliva pooled from 8–12 Ae. aegypti mosquitoes was cultured on LB agar plates for at least 5 days at 28 °C. Fungi appeared as grey–blue powdery or fuzzy colonies (indicated with black arrows, Fig. 1A), distinguishable from bacterial colonies. The fungi colony forming units (CFU) per mosquito were recorded after culturing for at least 120 h. No fungal growth was observed in the negative environmental control, which contained the blank salivation solution present on the bench during the collection of the saliva. Next, saliva collected from single Ae. aegypti and field-collected Cx. pipiens female mosquitoes was cultured in similar conditions. A total of 40% of Ae. aegypti (12/30 females) and 33% of Cx. pipiens (7/21 females) had culturable fungi in their saliva (Fig. 1B). ITS3-4 universal qPCR, which determined the fungal load, was performed to verify the presence of fungal DNA in saliva, salivary glands, and midgut samples of Ae. aegypti. The 17S mosquito housekeeping gene was used as an internal control. The highest fungal load was detected in midgut samples, with 4.5 × 10^6^ median ITS copies/mosquito (Fig. 1C). Salivary glands contained a fungal load of 3.0 × 10^3^ median ITS copies/mosquito, which was higher than the fungal load in saliva samples (83 median ITS copies/mosquito). The latter was not significantly different from the fungal load in the negative control (Kruskal–Wallis test).Fig. 1. Presence of fungi in the saliva of Aedes aegypti and Culex pipiens mosquitoes. A Mosquito saliva of Ae. aegypti (laboratory strain) was harvested by forced salivation and cultured on LB agar plates for 120 h at 28 °C. Fungal colonies are representatively indicated by black arrows. B Mosquito saliva from single Ae. aegypti (laboratory strain) and Cx. pipiens (field) mosquitoes was cultured on LB agar plates. The number of CFU/mosquito was quantified (by visual counting) after culturing for 120 h at 28 °C. Grey lines show the median value of the tested samples. The data were analyzed with the Kruskal–Wallis test, followed by Dunn’s multiple comparisons with correction. C Genome copies of fungi in different organs of Ae. aegypti were quantified by ITS qPCR. Negative control (NC) consisted of the blank solution used as environment control for forced salivation. Each point represents an independent pool containing samples harvested from 8–12 mosquitoes normalized per number of mosquito. Lines show the median value of the tested samples. LOQ presents the limit of quantification of the qPCR. Data were analyzed with the Kruskal–Wallis test, followed by Dunn’s multiple comparisons with correction (^**^P < 0.01). SG salivary glands. D Maximum likelihood tree was constructed using MEGA-X with the substitution model Kamura 2-parameter model (K2) and gamma distributed (G) (500 bootstrap replicates), by using the ITS3-4 segments of Penicillium crustosum identified in mosquito saliva (OL436123; isolated from Ae. aegypti and Cx. pipiens) and other fungal species from GenBank that were previously detected in mosquito body and organs
To identify the fungal colonies cultured from Cx. pipiens or Ae. aegypti saliva on agar plates, DNA was extracted from individual colonies and subjected to a universal ITS3–4 fungal identification PCR [32] and subsequent Sanger sequencing analysis. The results showed a 99% identity similarity by BLAST (NCBI) for both Penicillium expansum and Penicillium crustosum. Therefore, a specific PCR for Penicillium crustosum was performed [35]. An 892 bp segment could be amplified by P. crustosum-specific primers from all samples (Supplementary Fig. 1), indicating that the fungal colonies were Penicillium crustosum rather than Penicillium expansum.
A phylogenetic analysis was performed to compare the ITS3–4 segments of Penicillium crustosum detected in the mosquito saliva from this study to the Penicillium crustosum and Penicillium expansum species previously reported in GenBank that were detected in mosquito bodies and organs (Fig. 1D). The bootstrap values indicate how many times (out of 500 repeats) the sequences clustered together when repeating the tree construction on a resampled data set. In 89% of the possibilities, the Penicillium clade, including the species identified in the mosquito saliva, was related to the previously described mosquito-colonizing Penicillium chrysogenum [36]. In 97% of the possibilities, the Penicillium clade was related to a clade containing Aspergillus fumigatus, Aspergillus niger, and Aspergillus flavus, which were previously reported in mosquitoes [37].
Identification of bacteria in mosquito saliva
Culturing of mosquito salivary bacteria was performed on LB agar plates, as no significant difference was obtained when comparing the bacterial load and diversity of mosquito salivary bacteria using LB agar plates or BHI/blood agar plates (Supplementary Fig. 2 and Supplementary Tables 1 and 2). For the culturing, saliva from 8–12 mosquitoes was pooled, and 50 µL of the pool was inoculated on agar plates. In a first step, bacteria were characterized by morphological features (e.g., shape and color) (Fig. 2A, bacteria indicated with black arrows). The bacterial CFU/mosquito was defined at 48 h post culture at 28 °C for both Ae. aegypti and Cx. pipiens (Fig. 2B). No colonies grew in the environmental control, containing only the salivation solution placed on the bench during the salivation procedure. The bacterial CFU/mosquito for Cx. pipiens was significantly different from the environmental control (Kruskal–Wallis test, P < 0.05). To further support this, ‘salivation’ was also performed using dead Ae. aegypti females (previously killed at −20 °C for 5 min) or by cutting the proboscis of live females and immersing it in 20 µL of blank solution, the collected saliva was cultured under the same conditions, and no bacterial or fungal colonies were observed. Furthermore, no colonies were observed on the plates with salivary gland samples from the same mosquitoes (Supplementary Fig. 3). Using 16S qPCR, the highest bacterial load was found in the midgut of Ae. aegypti (10^4^ median copies/mosquito), followed by saliva (170 median copies/mosquito) and salivary glands (38 median copies/mosquito) (Fig. 2C). The latter was significantly different from the bacterial loads in midguts (P < 0.005; Kruskal–Wallis test). Individual bacterial colonies were picked from the plates on the basis of their characteristics (shape, color, and size) and were identified by 16S Sanger sequencing (Fig. 2D). Klebsiella spp., Enterobacter spp., * Asaia* spp., Serratia marcescens, Serratia nematodiphila, Microbacterium spp., and Chryseobacterium sp. were identified in the saliva of Ae. aegypti (shown with ○ in Fig. 2E and presented in Supplementary Table 1), whereas only Serratia marcescens and Serratia nematodiphila were found in the saliva of Cx. pipiens mosquitoes shown with ∆ in Fig. 2E and presented in Supplementary Table 2). Phylogenetic analysis showed that, in 98% of the possibilities, Serratia sp. sequences of this study clustered with Serratia sp. (FJ372764) previously found in mosquitoes (Fig. 2E) [38]. In addition, the clade containing Enterobacter spp. and Klebsiella spp. was related to those previously identified in the soil where the mosquitoes resided (OK035565 and OL441036) in 87% and 89% of the possibilities, respectively. In 74% of the possibilities, the Asaia spp. detected in saliva were closely related to the Asaia sp. that was previously detected in mosquitoes (FJ372761) [38]. Finally, the Chryseobacterium sp. and Microbacterium sp. were found in 100% of the possibilities related to the species previously observed in mosquitoes (KY002136 and OM033360).Fig. 2. Presence of bacteria in the saliva of Aedes aegypti and Culex pipiens mosquitoes. A Mosquito saliva of Ae. aegypti (laboratory strain) was harvested by forced salivation and cultured on LB agar plates. Bacterial colonies are representatively indicated by black arrows. B Mosquito saliva pools from both Ae. aegypti (laboratory strain) and Cx. pipiens (field collection) were cultured on LB agar plates at 28 °C. The CFU/saliva pool was quantified (by visual counting) after culturing for 48 h. Each dot represents one pool of 8–12 saliva samples normalized per number of mosquito. Lines show the median value of the tested samples. The data were analyzed with the Kruskal–Wallis test, followed by Dunn’s multiple comparisons with correction (^^P < 0.05). C Genome copies of the 16S bacterial gene in different organs of Ae. aegypti were quantified by 16S qPCR. NC as environmental control consisted of the solution used for forced salivation. Each point represents an independent pool containing samples harvested from 8–12 mosquitoes normalized per number of mosquito. Lines show the median value of the tested samples. Data were analyzed with the Kruskal–Wallis test, followed by Dunn’s multiple comparisons with correction (^**^P < 0.001). LOQ presents the limit of quantification of the qPCR. SG salivary glands. D Identification by 16S Sanger sequencing of the individual bacterial colonies (on the basis of shape, color, and size) detected on the agar plates after cultivating Ae. aegypti and Cx. pipiens saliva. E ML tree was constructed using MEGA-X with the substitution model being Kamura 2-parameter model (K2) and gamma distributed (G) (500 bootstrap replicates), by using the Sanger sequences of bacterial colonies identified in mosquito saliva (Ae. aegypti indicated with ○ and Cx. pipiens indicated with ∆) and other species from GenBank that were previously detected in mosquitoes
To profile the bacterial composition, 16S metagenomic analysis was performed on saliva, salivary gland, and midgut sample pools of Ae. aegypti females without a prior cultivation step. DNA extraction of the saliva samples resulted in a relatively low DNA yield (around 10 ng/µL). Approximately 10,000 reads were obtained per saliva pool, with a total read base of 70 Mbp/sample pool and Q30 of more than 82%. The sequencing outputs of saliva samples (1.1 × 10^5^ reads, standard deviation (SD) ± 1.24 × 10^4^) were equal or slightly higher than the ones of midguts (9.5 × 10^4^ reads, SD ± 1.3 × 10^4^) and salivary glands (1.1 × 10^5^ reads, SD ± 4.1 × 10^3^) (Supplementary Table 3). There was no significant systematic difference in alpha diversity among saliva, midgut, and salivary gland samples (Kruskal–Wallis chi-squared = 3.5043, df = 3, P = 0.3202) (Fig. 3A). However, ordination picked out a clear separation between the saliva samples and the other organ samples (Fig. 3B). The taxonomic distribution of the top 15 bacterial sequences of saliva, salivary glands, and midguts was generated at the genus level (Fig. 3C). Two dominant genera in salivary glands were Pseudomonas (55% ± 27%) and Cutibacterium (5% ± 3%). In saliva samples, genera appeared more diversely, with the most relatively dominant genera being Delftia (21% ± 18%), Methylobacterium–Methylorubrum (5% ± 3%), Flavobacterium (7% ± 5%), and Chryseobacterium (6% ± 5%). In midguts, the top three genera were Enterobacter (36% ± 14%), Pseudomonas (13% ± 4%), and Cutibacterium (11% ± 7%). Interestingly, the most dominant genus in the blank control was Enhydrobacter (19%), which was rarely found in the mosquito samples.Fig. 3. Bacterial richness and diversity in saliva, midguts, and salivary glands of Aedes aegypti mosquitoes. The 16S rRNA V3–V4 metagenomic sequencing was analyzed in DADA2. A alpha diversity in the microbial communities identified from different organs was visualized by Phyloseq and scored using the “observed” and “Simpson” diversity indexes; no significant systematic difference in alpha diversity among saliva, midgut, and salivary gland samples (Kruskal–Wallis chi-squared = 3.5043, df = 3, P = 0.3202). B Ordination was assessed using Bray–Curtis distances; C Taxonomic distribution of the top 20 sequences was shown at the genus level. SG salivary glands
Effect of blood feeding, life stages, and sex on the mosquito microbiota
To assess whether blood feeding could influence the presence of the saliva microbiota, culturable fungi and bacteria were determined in the saliva of Ae. aegypti following a blood meal. Our results showed that the bacterial and fungal loads were not different in the saliva of fed versus non-fed mosquitoes (Fig. 4A, B), demonstrating that a blood meal did not alter the microbiota loads in the saliva. Of note, no fungal or bacterial colonies were observed after 1 week of culture of the blood itself, indicating there were no input fungi or bacteria from the blood meal.Fig. 4. The effect of blood feeding and life stages on the microbiota. A Fungal and B bacterial CFU of saliva from 1-week-old female Ae. aegypti adults fed on rabbit blood. Saliva was harvested after 1 week and 2 weeks post blood meal. The CFU/saliva pool was quantified (by visual counting) after culturing. Each dot represents one pool of 8–12 saliva samples normalized per number of mosquito. Lines show the median value of the tested samples. The loads were compared between fed and unfed groups. Statistical analysis was performed using the Mann–Whitney test. C Genome copies of fungi at different life stages of Ae. aegypti were quantified by ITS qPCR using homogenized mosquito bodies. The lines represent the median values. LOQ presents the limit of quantification of the qPCR. D0: newly emerged adults. Negative control (NC) consisted of the blank solution used as environment control for forced salivation. Statistical analysis was performed using the Kruskal–Wallis test followed by Dunn’s multiple comparisons with correction (^^P < 0.05; ^**^P < 0.01). D Bacterial loads in different life stages of Aedes aegypti. The CFU read-out was performed by visual counting after culturing the homogenized mosquito bodies for 48 h at 28 °C on LB agar plates. The lines represent the median values. D0: newly emerged adults. Negative control (NC) consisted of the blank solution used as environment control for forced salivation. Statistical analysis was performed using the Kruskal–Wallis test followed by Dunn’s multiple comparisons with correction (^^P < 0.05)
The effect of the mosquito’s life stages on its microbiota in Ae. aegypti was determined with qPCR in larvae, pupae, and female/male adults (Fig. 4C, D). The fungal loads increased during the immature life stages from 3.3 × 10^3^ median ITS copies/mosquito in L1/L2 larvae to 3.0 × 10^5^ median ITS copies/mosquito in male pupae (Kruskal–Wallis test; P < 0.05.) and to 1.0 × 10^6^ median ITS copies/mosquito in female pupae (P < 0.01) (Fig. 4C). A drop in the fungal load was observed during the metamorphosis of male and female pupae into newly emerged male and female mosquitoes, although nonsignificant (P > 0.05). This initial drop was followed by an increase in fungal load in adult female mosquitoes, with a significant difference for 3-week and 4-week-old females compared with L1/L2 larvae (P < 0.05). There was no significant difference in fungal loads between adult males and females (P > 0.05).
In contrast to fungi, the bacterial loads in immature life stages were already high and did not increase further in the adult life stage. Indeed, no significant difference was observed between larvae or pupae compared with adult mosquitoes for the bacterial loads (Fig. 4D). A similar drop was observed during metamorphosis in bacterial genome copies from pupae to newly emerged adult mosquitoes, although nonsignificant. The bacterial load in female mosquitoes was higher compared with the loads in male mosquitoes, with a statistically significant difference in median bacterial colonies between 4-week-old females (CFU/mosquito of 1.8 × 10^5^) and 4-week-old males (1.5 × 10^3^ CFU/mosquito) (Kruskal–Wallis test, Dunn’s comparison; P < 0.05) (Fig. 4D). Supplementary Figure 4 shows the culturable bacteria identified by Sanger sequencing in Ae. aegypti at different life stages in males and females.
A correlation analysis was performed for male and female mosquitoes to investigate the relationship between bacterial and fungal loads for the adult life stages. Interestingly, an opposite correlation was observed in male compared with female mosquitoes. In male mosquitoes, a significant negative correlation was observed between bacterial and fungal loads (Pearson correlation; R^2^ = 0.3281; P < 0.05). This showed that male mosquitoes with a high bacterial load had lower fungal loads (Fig. 5A). This was in contrast to the significant positive correlation in females between bacterial and fungal loads (Pearson correlation; R^2^ = 0.1395; P < 0.05) (Fig. 5B). Female mosquitoes with a high bacterial load also had a high fungal load. The difference in correlation between male and female could be linked to the blood feeding-based physiology of females, although this requires further investigation.Fig. 5. Correlation between fungal and bacterial loads in males and females. A Genome copies of fungi in of Ae. aegypti (males and females); homogenized mosquito bodies were quantified by ITS qPCR. B Genome copies of the 16S bacterial gene of Ae. aegypti (males and females); homogenized mosquito bodies were quantified by 16S qPCR. Each point represents an independent pool containing samples harvested from 8–12 mosquitoes normalized per number of mosquito. Statistical analysis was performed using the Pearson correlation test
Depletion of culturable fungi and bacteria in mosquito saliva after oral treatment of adult mosquitoes
To obtain mosquito saliva devoid of fungi or bacteria, adult mosquitoes were given an oral antifungal or antibiotic treatment, respectively. To deplete fungi, female Ae. aegypti of 5–7 days old were provided with 10% sterile sugar solution supplemented with 25 µg/mL Fungin for 10 days (Fig. 6A). Antifungal treatment of the adult mosquitoes resulted in the complete absence of fungi in the saliva (Fig. 6A), whereas the bacterial load remained unaffected (Fig. 6C). To deplete bacteria, females were provided with 10% sterile sugar solution supplemented with 20 units/mL of penicillin and 20 µg/mL streptomycin for 6 days, as described previously [20]. Oral antibiotic treatment reduced, but did not completely eliminate, the bacterial load in the saliva (Fig. 6B). Remaining bacteria following this treatment were identified as C. cucumeris, M. maritypicum, M. oxydans, and S. maltophilia, representing 17%, 69%, 12%, and 2.7% of all colonies, respectively. Therefore, gentamycin was added to the antibiotic cocktail, a drug effective against both Gram-positive and Gram-negative bacteria. The addition of gentamycin resulted in a significant and complete depletion of culturable bacteria in the saliva (Fig. 6B) (P < 0.01). Different doses of penicillin, streptomycin, and gentamycin were evaluated to optimize the treatment (Fig. 6C). In addition, oral antifungal and antibiotic treatments at different doses were shown not to affect the survival of females (Supplementary Fig. 5). As it was possible to obtain saliva devoid of bacteria or fungi after orally treating the mosquitoes, we concluded that the culturable bacteria and fungi detected in saliva originated from the mosquito body itself and not from the environment or the mosquito outer body.Fig. 6. Depletion of culturable fungi or bacteria in mosquito saliva after oral treatment. A Fungal load in saliva from antifungal-treated mosquitoes (AF; 25 µg/mL of Fungin in 10% sucrose for 10 days) and nontreated saliva (NT). The CFU/saliva pool was quantified (by visual counting) after culturing. Each dot represents one pool of 8–12 saliva samples normalized per number of mosquitoes. B Bacterial load in saliva from antibiotic-treated mosquitoes (AB; 20 units/mL of penicillin and 20 µg/mL streptomycin in 10% sucrose for 6 days with and without 100 µg/mL gentamycin) and nontreated saliva (NT). The CFU/saliva pool was quantified (by visual counting) after culturing. Each dot represents one pool of 8–12 saliva samples normalized per number of mosquitoes. C Female Ae. aegypti adults (5–7 days old) were divided between three treatment groups: untreated control group (NT: only 10% sucrose), antifungal-treated (AF: 25 µg/mL of Fungin in 10% sucrose for 10 days), and antibiotic-treated (AB: serial dilution up to 200 units/mL of penicillin and 200 µg/mL streptomycin + gentamycin in 10% sucrose for 6 days). Bacterial loads in the individual bodies were quantified by determining CFU/mosquito. Lines show the median value of the tested samples. LOD presents the limit of detection. Significant differences were demonstrated by the Mann–Whitney test (two groups) or Kruskal–Wallis test (three groups or more, followed by Dunn’s multiple comparisons)(^^P < 0.05; ^^P < 0.01; ^^P < 0.001)
Reduced viral titers after co-incubation of Semliki Forest virus with saliva of antibiotic- or antifungal-treated mosquitoes
To investigate the potential impact of fungi and bacteria in the mosquito saliva on arbovirus replication in vitro, saliva was collected from antifungal-treated females (AF) or antibiotic-treated females (AB) and from nontreated female Ae. aegypti (NT) (Fig. 7A–C). In addition, filtered saliva from untreated mosquitoes, in which fungi and bacteria were removed physically using a 0.2-µm filter, was tested. Interestingly, the direct inoculation of human skin fibroblasts with SFV and AB saliva significantly reduced SFV replication compared with the NT saliva (Kruskal–Wallis test; P = 0.0457) (Fig. 7A). AF saliva also reduced SFV infection to a similar extent, but this reduction was not statistically significant (P = 0.105). An inhibition was not observed when skin fibroblasts were incubated with filtered saliva (Fig. 7A). Similarly, pre-incubation of saliva and SFV for 1 h prior to inoculation on human skin fibroblasts significantly lowered SFV replication for AB saliva or AF saliva compared with SFV and for AF saliva compared with NT saliva (Kruskal–Wallis test). Again, there was no difference induced by the filtered saliva (Fig. 7B). Furthermore, in both conditions, no difference was observed between SFV infectivity in the presence or absence of NT saliva. Finally, saliva or PBS was pre-incubated with skin fibroblast cells for 1 h, after which the cells were infected with SFV. No significant difference was observed between the NT saliva, AF saliva, or AB saliva conditions compared with the PBS control group (SFV); however, SFV replication was significantly reduced when cells were pre-incubated with filtered saliva (Kruskal–Wallis test; P = 0.0417) compared with the PBS control group (Fig. 7C).Fig. 7. Effect of mosquito saliva on Semliki Forest virus infectivity in human skin fibroblasts. SFV infectious titers following exposure to mosquito saliva. For the positive control (SFV group), saliva was replaced by 1× PBS. A Human skin fibroblasts were directly inoculated with saliva (saliva from five females/well) and SFV (MOI:0.001). B SFV (MOI:0.001) was pre-incubated with saliva for 1 h at 28 °C before inoculation on human skin fibroblasts. C Skin fibroblast cells were preincubated with saliva 1 h before infection with Semliki Forest virus (MOI: 0.001), and cells were washed three times. The cell supernatant was harvested at 24 h after infection, and the viral yield was determined by endpoint titrations on Vero cells. TCID_50_/ml was calculated using the Reed and Muench method. Kruskal–Wallis test followed by Dunn’s multiple comparisons with correction was used for statistical analysis (ns, P > 0.05; ^^P < 0.05; ^^P < 0.01; ^^P < 0.001). Only significant differences are shown in the figure; nonsignificant results from pairwise comparisons are not indicated to avoid overcrowding the figure. The dotted line indicates the limit of quantification (LOQ). NT saliva: saliva of untreated mosquitoes; AF saliva: saliva from mosquitoes treated with 25 µg/mL of Fungin in 10% sucrose for 10 days; AB saliva: saliva from mosquitoes treated with 20 units/mL penicillin, 20 µg/mL streptomycin, and 100 µg/mL gentamycin in 10% sucrose for 6 days
Discussion
Mosquito saliva is a complex mixture of protein, lipid, and microbial components; however, the latter has not been studied in great detail. Here, we aimed to explore the presence of bacteria and fungi in Aedes and Culex mosquito saliva. Most salivary bacteria identified in this study were previously reported in either the midgut or other mosquito organs (Fig. 2). Identified Gram-negative genera were Asaia, Enterobacter, Klebsiella, Chryseobacterium,, and Serratia. Interestingly, Asaia, belonging to the Acetobacteraceae family, was previously shown to colonize the salivary glands and the midgut of mosquito species such as Ae. aegypti, Aedes albopictus, Anopheles stephensi, and Anopheles gambiae [13, 39]. Importantly, Asaia has been considered a potential vector of antiplasmodial factors through a paratransgenic approach, where the symbiotic bacterium acts as a Trojan horse to produce components that target the pathogen [40]. The bacterial genera Enterobacter and Klebsiella, belonging to the family Enterobacteriaceae, are both frequently found in the gut of adult Aedes spp. and Cx. pipiens [16, 41]. Furthermore, Serratia of the Yersiniaceae family was identified in the salivary glands and the midgut of Aedes and Anopheles mosquitoes [13] and in the midgut of Culex mosquitoes [41]. Interestingly, Serratia marcescens was found to have the ability to render mosquitoes more permissive to arbovirus infection [20]. Chryseobacterium, a member of the Weeksellaceae family, was detected in all life stages of Ae. aegypti [42], which is in line with our observations that this species was found in both pupae and saliva samples of adult females. Microbacterium, belonging to the family Microbacteriaceae, was reported to be abundantly present in the larval stage of Ae. aegypti [43]. In this study, these Gram-positive bacteria (e.g., M. maritypicum and M. oxydans) were mainly identified in the pupal stage, but also in Ae. aegypti adults, suggesting that a set of bacteria such as Chryseobacterium and Microbacterium might be conserved in Ae. aegypti colonies, despite being maintained in different laboratories [44].
Our results on the saliva microbiota are in line with previous findings showing the presence of bacteria in saliva from Ae. albopictus and Anopheles mosquitoes [25, 26]. Intriguingly, the same bacterial genera were identified in the saliva of Ae. aegypti (Paea strain) reared in our laboratory as in the saliva of An. gambiae (G3 strain, MR4, MRA-112) and An. stephensi (Sind-Kasur Nijmegen strain) reared in a laboratory in Italy [45]. The 16S NGS data obtained in this study showed that the bacterial community in saliva samples was more diverse than in midgut samples. These findings are consistent with previous research [46], which showed that Ae. albopictus saliva harbored a richer microbial community compared with its midguts. To confirm the observations of this study and avoid possible biases in NGS results caused by background or kit-related contamination, future work should focus on systematically including extraction blanks and environmental controls.
Our study does not allow us to distinguish the effect of mosquito species (Aedes versus Culex mosquitoes) from their adaptation to the laboratory (field-collected versus laboratory-reared mosquitoes), as we only used Aedes laboratory mosquitoes and Culex field mosquitoes. As a follow-up, an in-depth comparison between field and laboratory mosquitoes of the same species (i.e., mosquitoes reared for an extended period under standard conditions versus those recently transferred from the field to the laboratory), as well as further examination of the effects related to mosquito species would be of interest. Notably, Gómez et al. [47] very recently compared the bacterial species in laboratory-reared and field-collected Ae. aegypti in Colombian populations, showing that Pseudomonas and Acinetobacter were present in both conditions and that field mosquitoes harbored a more diverse microbiota compared with laboratory mosquitoes [47].
In Aedes and Culex mosquitoes, fungal communities are mainly composed of Ascomycota (73–92%) and Basidiomycota (8–25%), including yeasts and filamentous species [48]. Several species of filamentous fungi (e.g., Aspergillus gracilis, Penicillium sp.) and yeasts (e.g., Candida sp.) were highly detected. These fungi were shown to play a role in the development, survival, and reproduction of mosquitoes [48]. Previously, Penicillium spp. were identified in other mosquito species or habitats such as the breeding water [49]. For instance, in Iran, Penicillium spp. were found to be the most abundant entomopathogenic fungi isolated and identified from larvae and breeding water of Anopheles and Culex mosquitoes [50]. In Brazil, Penicillium citrinum was identified in Aedes larvae obtained from natural and artificial breeding sites [51]. Penicillium chrysogenum was found in all life stages of Cx. pipiens in Egypt [52] and is particularly of interest, as P. chrysogenum isolated from the midgut of field Anopheles mosquitoes rendered the mosquito more susceptible to a Plasmodium infection via suppressing the mosquito’s innate immune system [53]. Here, we isolated and identified for the first time the fungus Penicillium crustosum from the saliva of both an Ae. aegypti laboratory strain and Cx. pipiens field-collected mosquitoes (Fig. 1). P. crustosum is a common blue mold, morphologically characterized by typically gray–blue–green conidia. This species has been frequently detected in fruits (e.g., peaches, apples, and pears), wheat, peanuts and other nuts, propolis, and food waste [54, 55]. It is a food-borne ubiquitous fungal species that produces mycotoxins such as roquefortine C and penitrems [56]. Interestingly, penitrems have demonstrated insecticidal activity against the large milkweed bug (Oncopeltus fasciatus) and the Mediterranean fruit fly (Ceratitis capitate) [57]. P. crustosum was also reported as the dominant species of the filamentous Penicillium spp. in extremely cold environments, in samples isolated from subglacial ice of arctic glaciers in Norway [58]. Symbiotic relationships between fungi and their hosts are well established in three model organisms ranging from mutualism (e.g., fungus Mortierella elongata and algae Nannochloropsis oceanica) [59] and commensalism (e.g., Saccharomyces cerevisiae or Candida albicans in the human digestive tract) [60] to parasitism (e.g., Beauveria bassiana in arthropods) [61]. For instance, the pathogenic fungus Beauveria bassiana can utilize a cross-kingdom small-RNA effector to attenuate host immunity, thereby facilitating its infection in mosquito cells [62] or directly in insecticide-resistant Ae. aegypti mosquitoes [63]. Although antifungal treatment did not alter mosquito lifespan in this research, it would therefore be interesting to investigate further whether Penicillium crustosum and its mycotoxins affect mosquitoes’ other life history traits, vector competence, or downstream viral infection in the mammalian host. In this study, only one fungal species was identified by culture. In future studies, next-generation sequencing could be applied to characterize the broader fungal community, followed by targeted isolation using selective media such as potato dextrose agar and malt extract agar.
To date, there is a growing understanding of microbiota–arbovirus interactions and an expanding interest in using the microbiota for vector control and arbovirus outbreak containment. During a mosquito bite, saliva and virus are inserted simultaneously into the skin of the host [64]. Therefore, a thorough investigation into saliva microbiota and their potential contributions to critical stages in mosquito-borne virus infections is warranted. To investigate this, the presence of fungi in mosquito saliva needs to be eliminated. We obtained this by treating adults with the antifungal drug Fungin via the sugar meal. Depletion of the bacteria from the mosquito saliva was obtained using an oral treatment with an antibiotic cocktail consisting of gentamycin, penicillin, and streptomycin. Although this did not fully clear the bacteria from the entire mosquito body, we could obtain complete depletion of culturable bacteria in the mosquito saliva (Fig. 6).
Next, we aimed to investigate whether the saliva microbiota of Ae. aegypti interacts with viral particles in the mosquito saliva. Co-incubation of saliva from antibiotic- or antifungal-treated mosquitoes with SFV reduced the infectious virus load in human cells (Fig. 7A). Also, pre-incubation of mosquito saliva from treated mosquitoes together with the virus before infecting cells lowered the amount of infectious virus (Fig. 7B). In contrast, in both conditions, filtered saliva had no effect on viral infectivity compared with saliva from untreated control mosquitoes, suggesting that the physical presence of the saliva microbiota had no effect on infectivity. Moreover, the antiviral effect observed with saliva from antibiotic-treated mosquitoes is likely not due to presence of remaining bacterial secretions, since these secretions are also still present in the filtered saliva. We therefore hypothesize that the oral antibiotic and/or antifungal treatment affected the composition of saliva components. Another potential explanation could be that the orally ingested antibiotic or antifungal is carried over in the saliva, affecting virus infectivity.
In the cell pretreatment condition (Fig. 7C), the incubation with NT saliva resulted in a downward trend in SFV titers, although this reduction was not significant. In contrast to the other incubation conditions, AB saliva did not result in a reduction in viral titers when cells were pretreated, whereas filtered saliva did reduce viral infectivity. This suggests an effect of bacterial secretions on the cells. The results of NT and AB saliva on pretreated cells are similar to prior research showing that mosquito saliva slightly diminished SFV replication in skin fibroblasts [30]. The authors hypothesized that the presence of the salivary bacteria activated immune responses in the skin cells, leading to a higher viral resistance. However, in the pre-incubation with virus condition, we did not observe a similar reduction by mosquito saliva (from untreated Ae. aegypti) as seen in [30]. This discrepancy could be due to the use of mosquitoes with different microbiota communities (Ae. aegypti Liverpool strain versus Ae. aegypti Pacific Paea strain) or different virus strains (SFV constructed from plasmid as per [65] versus SFV Vietnam strain). Moreover, our study used a lower MOI for infection (0.001 versus 0.01) and a longer pre-incubation period of virus with saliva (1 h versus 20 min). Importantly, it has been equally shown that the virus-enhanced infection phenotype by mosquito saliva observed in mouse models could not be recapitulated in vitro or ex vivo [30]. Therefore, it is imperative to conduct in vivo studies to elucidate the potential effects of salivary bacteria and fungi on virus infection and pathogenesis.
Conclusions
This study detected fungi and bacteria in mosquito saliva by culturing saliva from two distinct mosquito species: Ae. aegypti and Cx. pipiens. Furthermore, 16S metagenomics analysis showed that the bacterial community in saliva was more diverse than in midguts. The application of antibiotics or an antifungal drug in the oral treatment of adult mosquitoes depleted microbiota in the saliva, indicating that the origin of saliva microbiota lies within the mosquito body. Moreover, the depletion of fungi and bacteria from saliva may influence resistance to viral infection in vitro. However, the impacts of saliva microbiota warrant further investigation in vivo. Collectively, this work lays the foundation for further investigation into the effect of saliva fungi and bacteria on arbovirus replication and infection.
Supplementary Information
Supplementary material 1: Fig S1: Gel electrophoresis after PCR amplification. Fig S2: CFU of salivary bacteria by culturing on different agar plates. Fig S3: Bacterial loads of individual midgut and salivary glands of Culex pipiens and Aedes aegypti mosquitoes. Fig S4: Bacteria composition in mosquitoes variation according to life stage and sex. Fig S5: Survival rate of mosquitoes recorded for each treatment group. Table S1: Bacteria identified in the saliva of Aedes aegypti. Table S2: Bacteria identified in the saliva of Culex pipiens. Table S3: Sequencing input and output reads used for downstream analysis.