Immune Myositis Complicating Follicular Lymphoma: Case Report
George Sarin Zacharia, Saran Lal Ajai Mokan Dasan, Chinazor Iwuaba

TL;DR
A rare case of follicular lymphoma presenting as inflammatory myopathy is reported, highlighting the importance of considering lymphomas in myopathy diagnoses.
Contribution
This case report adds to the understanding of follicular lymphoma's rare presentation as inflammatory myopathy.
Findings
A patient with follicular lymphoma presented with symptoms of inflammatory myopathy.
Treatment with methotrexate and rituximab led to significant symptom improvement.
The case underscores the diagnostic challenges of lymphomas mimicking other diseases.
Abstract
Background and Clinical Significance: Idiopathic inflammatory myopathies are a heterogeneous group of autoimmune disorders that may present as paraneoplastic syndromes. Although most frequently associated with solid-organ malignancies, hematological neoplasia, particularly lymphomas, is also likely linked. Case Presentation: We describe a sexagenarian female with progressive proximal muscle weakness, myalgias, and lymphadenopathy. Laboratory evaluation revealed markedly elevated creatine phosphokinase and myositis-specific antibodies: anti-Mi-2α and anti-EJ. Magnetic resonance imaging of the thighs confirmed active myositis. Lymph node biopsy reported follicular lymphoma. The patient was initiated on methotrexate and rituximab, with which she reported significant symptomatic relief. Conclusions: Inflammatory myopathy is an exceedingly rare presentation of follicular lymphoma. This case…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
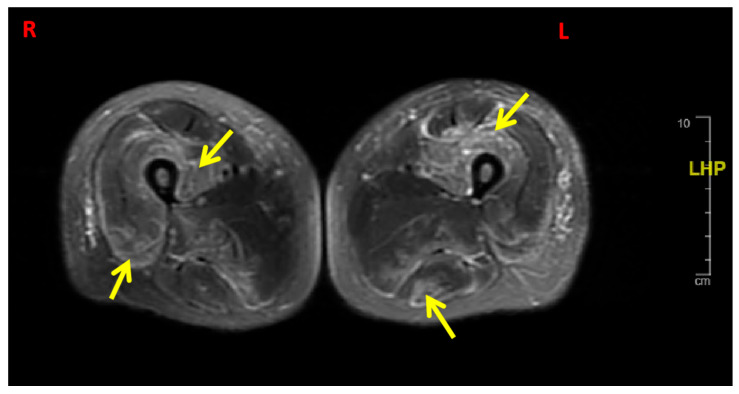 Figure 1
Figure 1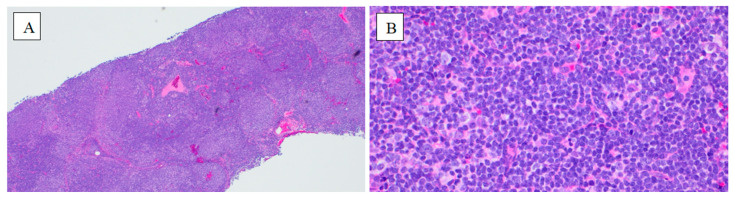 Figure 2
Figure 2Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsInflammatory Myopathies and Dermatomyositis · Parkinson's Disease and Spinal Disorders · Muscle and Compartmental Disorders
1. Introduction and Clinical Significance
Idiopathic inflammatory myopathies (IIM) are rare immune-mediated disorders with skeletal muscle inflammation and variable extramuscular involvement. Dermatomyositis and polymyositis are the classical subtypes; others include immune-mediated necrotizing myopathy, antisynthetase syndrome, and inclusion body myositis. Intriguingly, one-fourth of patients with adult-onset IIM are diagnosed with cancer within three years of diagnosis of IIM [1]. The link between myopathy and cancer likely involves shared autoantigens and dysregulated immune responses. Solid tumors, including ovarian, lung, and gastrointestinal cancers, are most frequently implicated. Hematological malignancies are less often associated with IIM. A retrospective study by Marie et al. identified 32 cases of dermatomyositis (DM) or polymyositis (PM) associated with hematological neoplasia over 5 years across 10 institutions; only one patient had follicular lymphoma [2]. The association of follicular lymphoma with IIM is exceedingly rare, with only a handful of cases published in the medical literature to date [2,3]. Here, we report a case of adult-onset inflammatory myopathy associated with follicular lymphoma, to highlight an uncommon clinical presentation and add to the published literature on the association between these pathological entities.
Clinical Significance: Adult-onset inflammatory myopathies are often paraneoplastic and are frequently associated with solid-organ neoplasia, whereas hematological malignancies are less common culprits. This case of inflammatory myopathy associated with follicular lymphoma is a rarity. Still, it highlights that lymphomas are ‘great masquerades’ and should be excluded in virtually all clinical presentations, especially when the etiology is unclear.
2. Case Presentation
A 61-year-old African American female presented with progressive proximal muscle weakness, generalized myalgias, and dysphagia to both solids and liquids of 2 months duration. She reported difficulty performing daily tasks such as combing her hair and rising from a seated position. Also, she noticed a weight loss of approximately 15 pounds over the same period. She denied fever, rash, Raynaud’s phenomenon, or arthralgia. Her past medical history was significant for hypertension, uterine fibroids, and diabetes mellitus.
Physical examination revealed proximal muscle weakness: grade 3/5 in the upper extremities and grade 4/5 in the lower extremities, respectively. There was no distal muscle weakness or sensory loss. She had palpable non-tender inguinal lymphadenopathy. No cutaneous lesions were identified, and the remainder of the systemic examination was unremarkable.
Laboratory investigations revealed significantly elevated creatine phosphokinase (10,510 IU/L), aspartate aminotransferase (400 IU/L), lactate dehydrogenase (912 IU/L), and aldolase (68.1 IU/L) levels. The baseline hematological and biochemical parameters are summarized in Table 1. She was positive for antinuclear antibody (ANA) with a titer of 1:1280, with a homogeneous/speckled pattern. The myositis-specific antibody panel was notable for the presence of anti-Mi-2α and anti-EJ antibodies, suggesting an inflammatory myopathy. Other autoimmune markers, including anti-dsDNA, anti-RNP, SSA/SSB, and Scl-70, were non-reactive.
Magnetic resonance imaging (MRI) with contrast of the thighs, performed on day 4 of initial presentation, revealed diffuse, symmetric muscle edema and enhancement throughout all compartments, which is consistent with active myositis (Figure 1). Abdominal ultrasound imaged fatty hepatomegaly, cholelithiasis, and multiple masses in the midline abdomen, which is most consistent with lymphadenopathy. Computed tomography of the chest, mammography, and echocardiography were within normal limits. The electromyography on day 16 was reported as short-duration, short-amplitude muscle unit action potentials and spontaneous fibrillation potentials, consistent with inflammatory myopathy.
A core biopsy of a right groin lymph node, performed on day 10 and reported on day 28 of initial presentation, confirmed follicular lymphoma, grade 1–2 of 3, with immunohistochemistry showing positivity for CD20, CD10, BCL-2, BCL-6, and PAX-5. The Ki-67 proliferation index was low (1–2+ out of 4+). Flow cytometry of peripheral blood and lymph node aspirate identified a monoclonal B-cell population (9%) that expresses CD19, CD20, CD10, and CD200, and lacks CD5 and CD23, which is consistent with a follicle center B-cell lymphoproliferative disorder. (Figure 2). Positron emission tomography (PET) demonstrated extensive hypermetabolic lymphadenopathy in the cervical, thoracic, abdominal, and pelvic regions, along with diffuse splenic involvement. A muscle biopsy was deferred as the patient opted against it.
The diagnosis of inflammatory myopathy associated with follicular lymphoma (Ann Arbor stage III; FLIPI score 5) was made, though it was limited by the absence of a muscle biopsy. The patient was initiated on methotrexate 15 mg weekly with folic acid supplementation once the diagnosis was made (day 18), resulting in significant symptomatic relief and a marked reduction in CPK levels. With regard to management of lymphoma, the oncology team, after extensive discussion with the patient, opted for rituximab monotherapy, as she declined cytotoxic chemotherapy. She was planned for four weekly doses followed by maintenance therapy. She received the first dose of rituximab almost 2.5 months after the initial presentation, at a dose of 375 mg/m^2^. The initiation of treatment was delayed as the patient was very hesitant to initiate any form of cancer therapy. She completed three doses of rituxumab and was subsequently lost to follow-up. At the last visit, with our institution, the patient continued under joint care by the rheumatology and oncology teams. As per clinic records, her symptoms resolved, and she was able to perform day-to-day activities independently. The serial laboratory parameters are summarized in Table 1.
3. Discussion
IIM, often referred to as myositis, is a heterogeneous family of autoimmune diseases that affects not only skeletal muscle but also other organ systems, such as the skin, joints, and lungs [4]. The estimated annual incidence and prevalence of IIM are 0.2–2 and 2–25 per 100,000, respectively [5]. They include DM, PM, immune-mediated necrotizing myopathy (IMNM), and inclusion body myositis (IBM). DM is the most common variant, while PM is the least common; IBM is the most common myopathy in individuals over the age of 50 years [6]. Genetic influences, molecular mimicry, and viral triggers play a potential role in the pathogenesis of IIM [7,8].
DM/PM presents with progressive, symmetric proximal muscle weakness developing over weeks to months. Distal weakness occurs and develops later in the course of the disease. Patients struggle with activities such as climbing stairs, rising from a chair, or combing hair. Facial and extraocular muscles are typically spared, while patients could have dysphagia [9]. DM is characterized by skin rashes such as heliotrope rash, Gottron’s papules, V-sign, and shawl sign. DM variants include amyotrophic DM, skin disease without overt muscle weakness, which still shows subclinical muscle involvement on biopsy, and ‘DM sine dermatitis’ with isolated muscle involvement. Contrary to DM/PM, IBM is frequently associated with early-onset distal weakness [6]. Clinical features of concomitant autoimmune diseases or underlying neoplasia may overlap with the clinical features of myopathy.
Diagnosis relies on markers of muscle injury in the presence of autoantibodies, supported by imaging features of myositis, electromyography (EMG), and biopsy. The muscle biopsy remains the gold standard for diagnosis and demonstrates inflammatory infiltrations. The serum markers of muscle injury include CPK, aminotransferases, lactate dehydrogenase, and aldolase. A myriad of autoantibodies have been described in IIM and cancer-associated myopathies; specific assays would help in diagnosis. Table 2 summarizes the types of autoantibodies and corresponding inflammatory myopathies. A large-scale study by Kerola et al., including more than 1000 subjects with positive myositis antibodies, found that anti-HMGCR antibodies had the highest positive predictive value (94%) for myositis. Antisynthetase antibodies had the highest positive predictive value for concurrent interstitial lung disease, while anti-TIF1-γ was best predictive of malignancies [10]. Anti-aminoacyl-tRNA synthetase (anti-ARS) antibodies are positive in 25–35% of patients with IIM, with anti-Jo-1 being the most common. This class of antibodies is most frequently associated with PM, interstitial lung diseases, and mechanic’s hands. Anti-Mi-2 antibodies are found in 10–30% of patients with IIM, typically DM. Also, anti-Mi-2 antibodies correlate with skin manifestations and lung sparing [11]. EMG reveals pathological spontaneous activity, which could be fibrillations, high-frequency discharges, or myotonic discharges. During voluntary muscle activity, owing to loss of myofibrils and contraction synchrony, patients with active myositis reveal small amplitude, short duration, and polyphasic motor unit potentials [12].
Adult-onset IIM, except for verified cases of IBM, is frequently associated with malignancies [1]. Inflammatory myopathies occur in 10–30% of all malignancies [3]. Solid-organ cancers: lung, gastrointestinal, breast, ovarian, pancreatic, and prostatic cancers are most frequently linked to inflammatory myositis. Histologically, adenocarcinomas account for almost 70% of myositis associated with neoplasia [13]. DM is the most frequent myositis associated with cancers. Results from the MYONET registry identified DM in 17% compared to 3% with antisynthetase syndrome, among those with cancer-associated myositis [14]. Hematological malignancies are less frequently associated with inflammatory myopathies, and those that are most frequently reported are the B-cell, non-Hodgkin’s lymphomas (NHL). In a single-center retrospective cohort study by Mecoli et al., IIM patients reported a standardized prevalence ratio of 2.71 for lymphoma overall and 3.91 for DM [15]. Follicular lymphoma, a type of B-cell NHL, is rarely associated with autoimmune or paraneoplastic syndromes. A literature review identified only a few cases of follicular lymphoma-associated myositis published to date [2,3,16].
Molecular mimicry between tumor and muscle antigens may lead to cross-reactive immune responses. Tumor neoantigens or stress-induced overexpression of these proteins can trigger both antitumor activity and myositis. The PD-1/PD-L1 pathway is dysregulated: elevated PD-L1 appears paradoxically protective in myositis, but persistent immune activation may overwhelm this effect, permitting both cancer growth and muscle inflammation [7].
Cancer-associated myositis typically demonstrates a temporal relationship with the neoplasm, resistance to standard therapies, improvement with cancer therapy/remission, and relapse with cancer recurrence [17]. Many studies classify inflammatory myopathy as cancer-associated myositis when cancer is diagnosed within three years of myositis diagnosis [18]. A 2018 study reported a median interval between diagnosis of malignancy and myositis as 1 month [19]. Zhou et al., in China, reported a median of 7.2 months between malignancy and myositis with an interquartile range of 0 to 20.4 months [20]. New onset inflammatory myopathy after the age of 40 years, dysphagia, DM, severe skin involvement, persistent disease activity despite immunosuppressive therapy, and anti-TIF1- or anti-NXP2 γ positivity are considered high risk for IIM related to cancer [1]. Except for patients with juvenile onset IIM or confirmed inclusion body myositis, all others with IIM are recommended to undergo basic cancer screening at the time of IIM diagnosis. In those with a high risk, a more comprehensive cancer screening is recommended [1].
Treatment of IIM includes addressing the underlying cause and using immunosuppression. Steroids are often considered the first-line agent. Methotrexate or azathioprine is frequently used as a steroid-sparing agent. Alternate agents, with limited data, include intravenous immunoglobulin, mycophenolate mofetil, rituximab, alemtuzumab, and cyclophosphamide [21]. Patients with cancer-associated myositis often respond to cancer chemotherapy and have a response correlated with tumor treatment response.
Our patient presented with proximal muscle weakness, dysphagia, and elevated muscle injury markers consistent with myositis, further supported by the radiological evidence and EMG pattern. She was positive for anti-Mi-2α and anti-EJ antibodies. Anti-Mi-2α is strongly associated with DM with a high positive predictive value and specificity [22]. However, the patient had isolated muscle involvement with no cutaneous lesions. Anti-EJ antibodies are anti-ARS autoantibodies typically associated with antisynthetase syndrome. Antisynthetase syndrome is characterized by myositis, interstitial lung disease, arthritis, and, less commonly, rashes, Raynaud’s phenomenon, and fever. It is considered a distinct subtype of IIM by some authors [23]. Anti-ARS are also frequent in PM, but anti-Jo-1 is the most frequent [24]. The concomitant presence of anti-Mi-2α and anti-EJ suggests the likelihood of an overlap syndrome, rather than an isolated pathology. She was confirmed to have NHL—follicular lymphoma on lymph node biopsy. Inflammatory myopathies associated with hematological malignancies are rare. Her myopathy symptoms responded well to immune modulation with methotrexate; however, her muscle enzymes, through a shown response, failed to normalize. She was hesitant to undergo lymphoma chemotherapy and ultimately decided to proceed with isolated monoclonal anti-CD20 therapy. Initiation of monoclonal antibodies further improved her CPK levels; however, she failed to complete the regimen and was lost to follow-up. In summary, this case depicts a probable paraneoplastic-triggered overlap myositis: a unique combination of follicular lymphoma with overlap myositis, PM, antisynthetase syndrome, or DM sine dermatitis.
Limitation: The lack of a muscle biopsy limits confirmation of the diagnosis. However, the combination of clinical features, imaging, and electrodiagnostic studies was highly suggestive of an inflammatory myopathy. As the patient was lost to follow-up, we are not able to provide the long-duration response/results to therapeutic interventions.
4. Conclusions
Inflammatory myopathies associated with follicular lymphoma are sporadic and diagnostically challenging. Adult-onset inflammatory myopathy should prompt screening for cancers. Clinicians should maintain a high index of suspicion for occult malignancies, including hematological neoplasia in patients with inflammatory myopathies.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Oldroyd A.G.S. Callen J.P. Chinoy H. Chung L. Fiorentino D. Gordon P. Machado P.M. Mc Hugh N. Selva-O’callaghan A. Schmidt J. International Guideline for Idiopathic Inflammatory Myopathy-Associated Cancer Screening: An International Myositis Assessment and Clinical Studies Group (IMACS) initiative Nat. Rev. Rheumatol.20231980581710.1038/s 41584-023-01045-w 37945774 PMC 10834225 · doi ↗ · pubmed ↗
- 2Marie I. Guillevin L. Menard J.-F. Hatron P. Cherin P. Amoura Z. Cacoub P. Bachelez H. Buzyn A. Le Roux G. Hematological malignancy associated with polymyositis and dermatomyositis Autoimmun. Rev.20121161562010.1016/j.autrev.2011.10.02422079677 · doi ↗ · pubmed ↗
- 3Melody M. Parrondo R. Moustafa M.A. Wu K. Menke D. Seim L. Tun H.W. Follicular lymphoma associated paraneoplastic myositis Clin. Case Rep.202082003200610.1002/ccr 3.304633088539 PMC 7562836 · doi ↗ · pubmed ↗
- 4Lundberg I.E. Fujimoto M. Vencovsky J. Aggarwal R. Holmqvist M. Christopher-Stine L. Mammen A.L. Miller F.W. Idiopathic inflammatory myopathies Nat. Rev. Dis. Primers 202178610.1038/s 41572-021-00321-x 34857798 · doi ↗ · pubmed ↗
- 5Khoo T. Lilleker J.B. Thong B.Y.-H. Leclair V. Lamb J.A. Chinoy H. Epidemiology of the Idiopathic Inflammatory Myopathies Nat. Rev. Rheumatol.2023969571210.1038/s 41584-023-01033-037803078 · doi ↗ · pubmed ↗
- 6Mantegazza R. Bernasconi P. Inflammatory Myopathies: Dermatomyositis, Polymyositis, and Inclusion Body Myositis Madame Curie Biosci. Database 2013 Available online: https://www.ncbi.nlm.nih.gov/books/NBK 6196/(accessed on 18 October 2025)
- 7Pu T. Sun J. Ren G. Li H. Neuro-immune crosstalk in cancer: Mechanisms and therapeutic implications Signal Transduct. Target. Ther.20251013310.1038/s 41392-025-02241-840456735 PMC 12130251 · doi ↗ · pubmed ↗
- 8Patasova K. Lundberg I.E. Holmqvist M. Genetic Influences in Cancer-Associated Myositis Arthritis Rheumatol.20227515316310.1002/art.4234536053262 PMC 10107284 · doi ↗ · pubmed ↗