Expansion of the Phenotype of Lymphatic Anomalies Caused by Somatic Activating BRAF Variant
Michael D. Fox, Sumukh Kumar, Allison D. Britt, Abhay S. Srinivasan, Lea F. Surrey, Seth E. Vatsky, Alexandra J. Borst, Hakon Hakonarson, Dong Li, Sarah E. Sheppard, Kristen M. Snyder, Denise M. Adams

TL;DR
This study expands the known symptoms of a genetic mutation in BRAF that causes lymphatic issues, highlighting the need for careful monitoring and treatment.
Contribution
The study expands the phenotypic spectrum of BRAF p.V600E-related lymphatic anomalies and highlights post-surgical conduction abnormalities.
Findings
Six individuals with complex lymphatic anomalies were found to have the BRAF p.V600E variant.
Abnormal lymphatic conduction was observed in five patients, often appearing years after surgery.
Phenotypic heterogeneity was noted among the cohort, emphasizing the need for longitudinal follow-up.
Abstract
The somatic activating variant in BRAF (p.V600E) was recently described as a novel cause of macrocystic head and neck lymphatic malformations in three individuals. Other recent studies profiling the genetic causes of more complex lymphatic anomalies identified this same pathogenic BRAF variant. Our aim was to expand the phenotypic description of the somatic BRAF p.V600E variant in individuals with vascular anomalies. We searched the database of individuals with vascular anomalies at our institution for those identified as having complex lymphatic anomalies and somatic BRAF p.V600E variants. A comprehensive retrospective review of identified individuals’ electronic health records was conducted. Six individuals with complex lymphatic anomalies had the BRAF p.V600E variant. All individuals had diffuse lymphatic malformations and abnormal lymphatic conduction. The conduction abnormalities…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
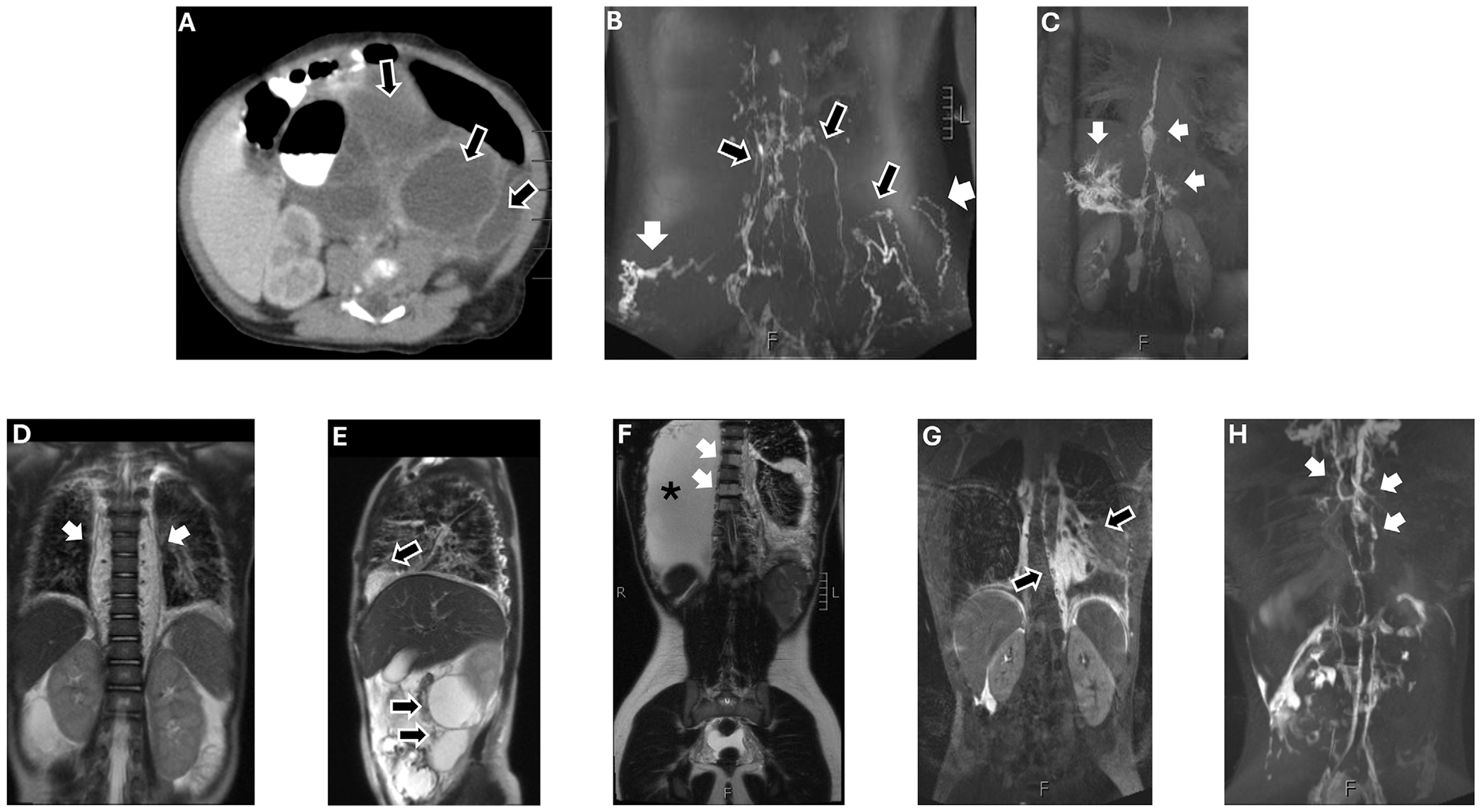 Figure 1
Figure 1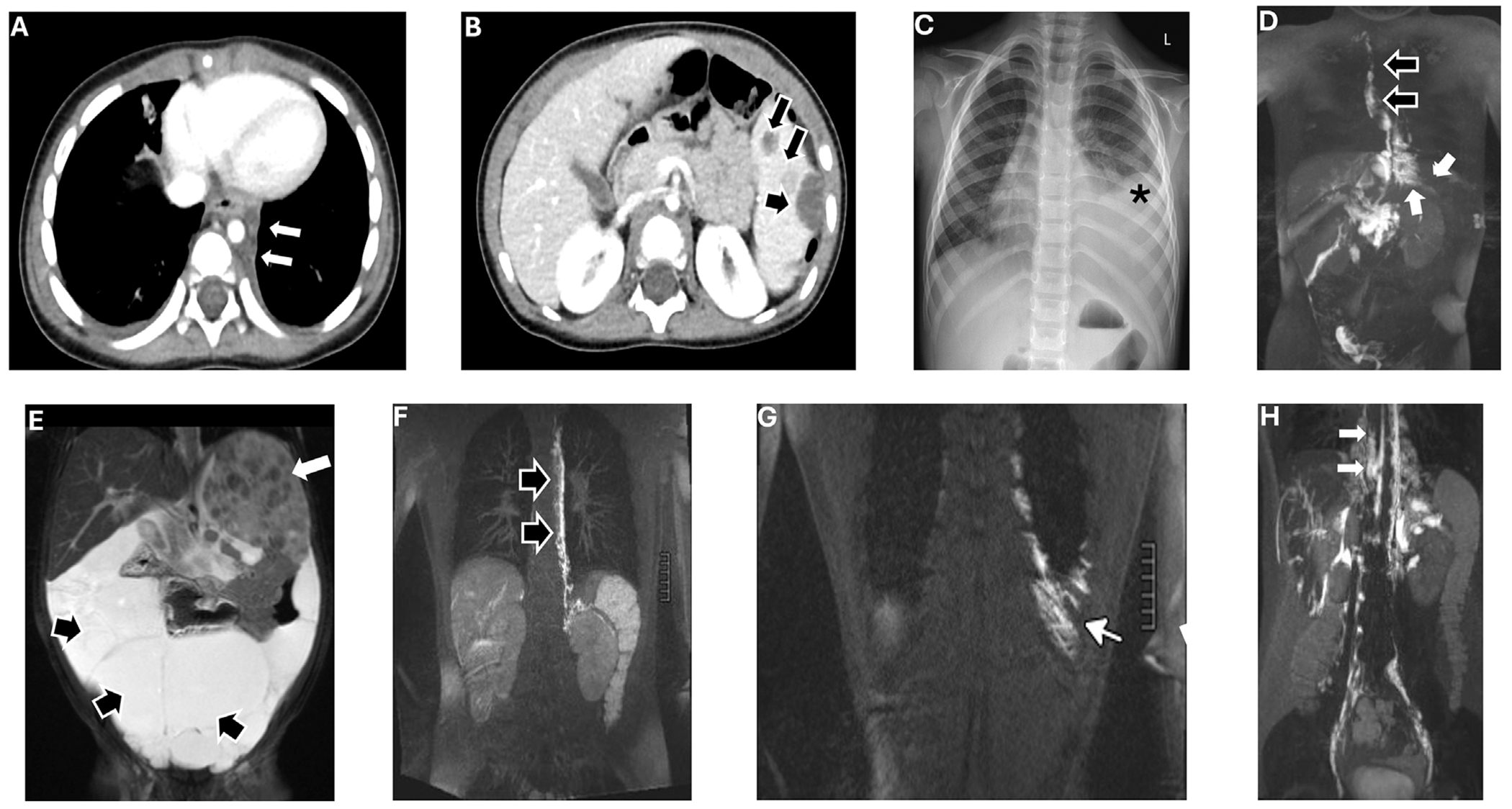 Figure 2
Figure 2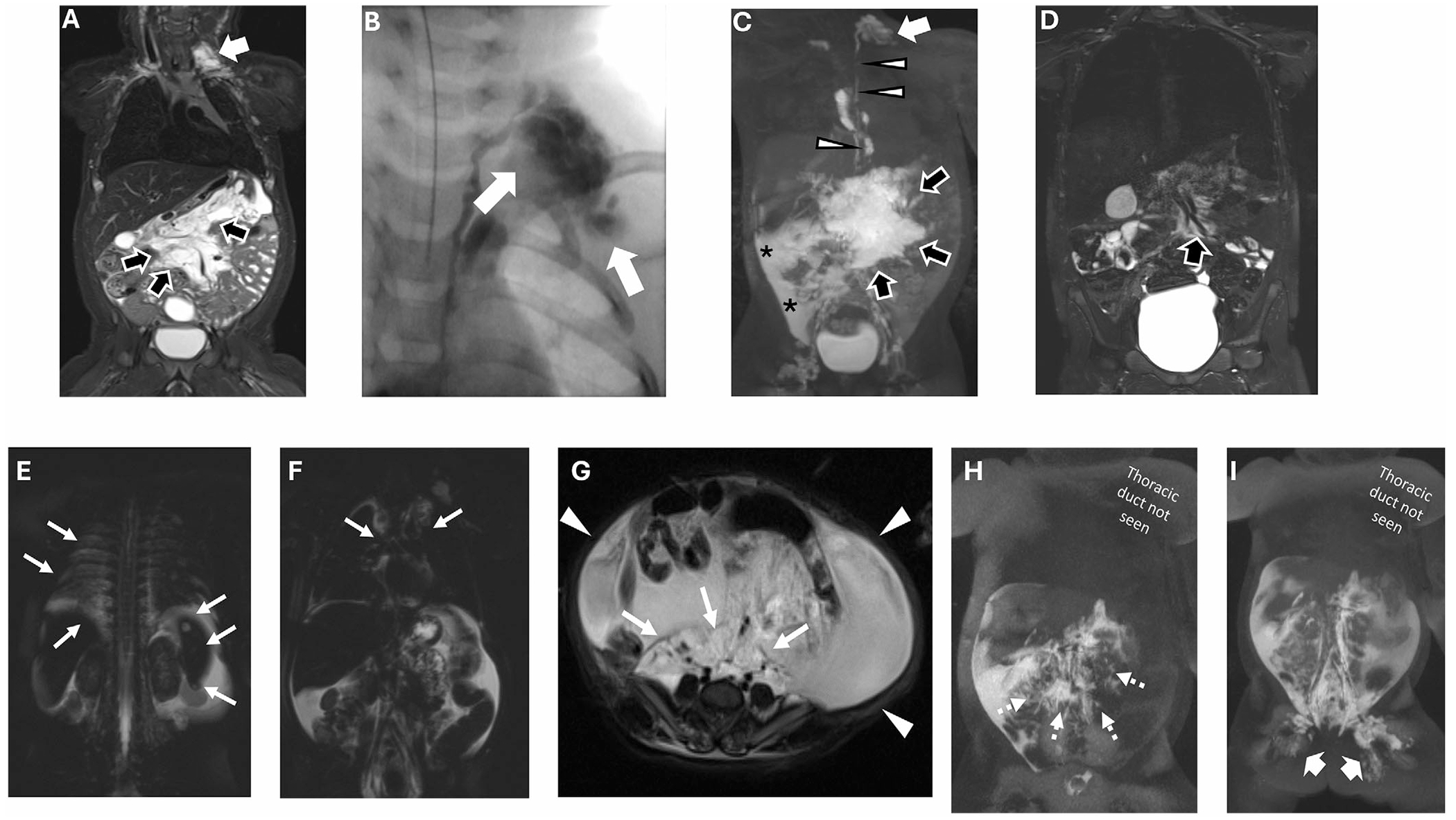 Figure 3
Figure 3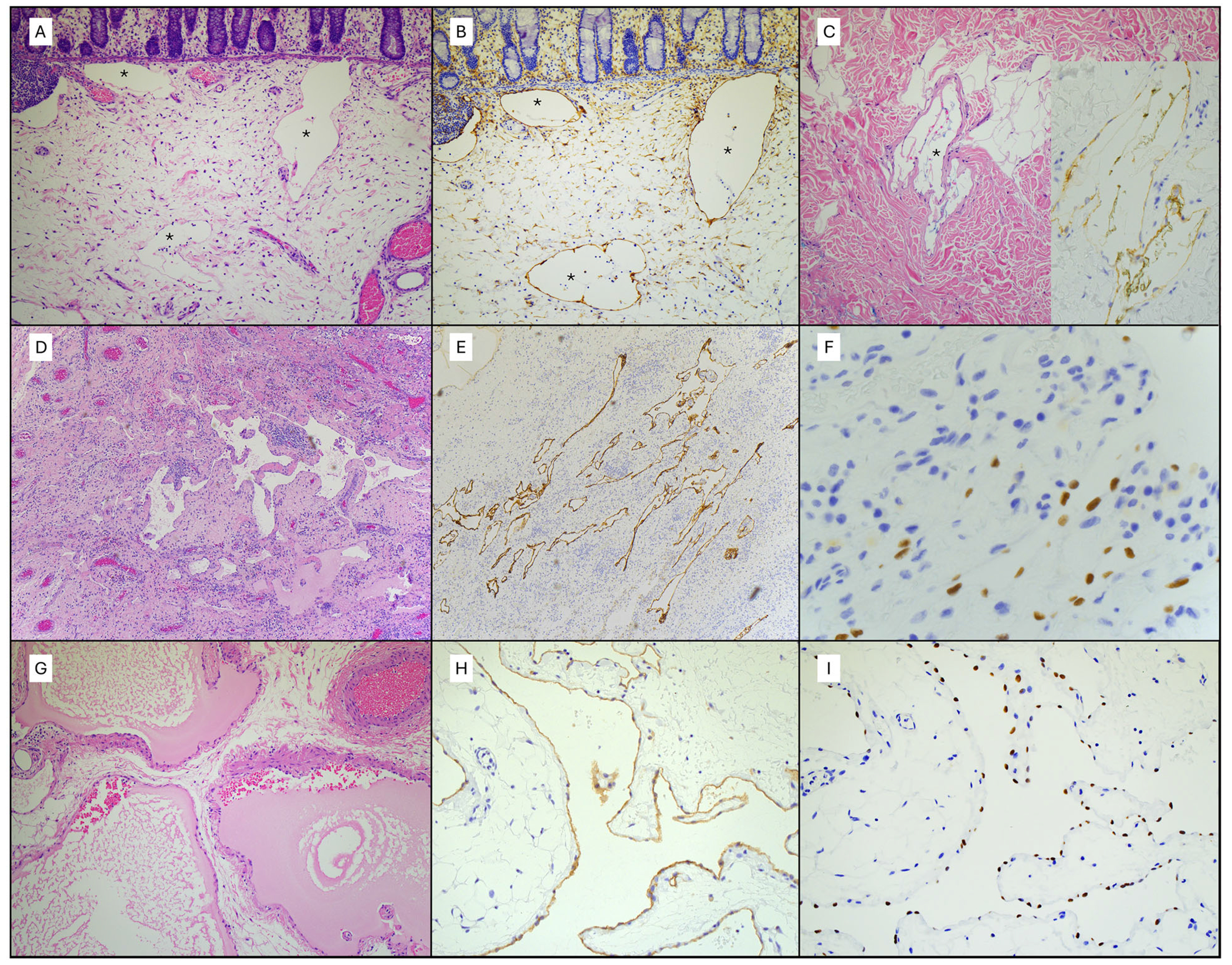 Figure 4
Figure 4Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsHedgehog Signaling Pathway Studies · RNA regulation and disease · Neurofibromatosis and Schwannoma Cases
Introduction
1 |
Complex lymphatic anomalies (CLA), which include generalized lymphatic anomaly (GLA), kaposiform lymphangiomatosis (KLA), Gorham Stout disease (GSD), and central conducting lymphatic anomaly (CCLA), are a highly morbid group of disorders characterized by abnormal development of lymphatic vessels resulting in diffuse lymphatic malformations (LM) and impaired lymphatic flow manifesting as effusions. Specific diagnosis of a CLA is made based on a combination of clinical presentation, imaging features, and histopathology (though often this is not available), but can be complicated by broad phenotypic heterogeneity as well as overlapping clinical features within this group of disorders [1–3].
GLA is notable for multifocal LM involving the soft tissues, thoracic and abdominal viscera and cavities, and bone. The bony involvement is often noncontiguous and does not result in destruction of the bone cortex. Affected tissue demonstrates dilated lymphatic channels that stain positive for D2-40 and PROX-1 (markers of lymphatic endothelium) [1–4]. KLA is similar to GLA in its areas of involvement, but commonly also involves coagulopathy with hemorrhagic effusions, and tends to have greater morbidity and higher risk of cardiopulmonary compromise due to its progressive and infiltrative nature. Individuals with KLA frequently have markedly elevated levels of angiopoietin-2, and histopathology demonstrates spindled lymphatic endothelial cells [1–5]. GSD, or “vanishing bone disease,” classically involves the bone in a contiguous pattern and results in progressive osteolysis with destruction of the bone cortex, but may also involve soft tissues and viscera. Evidence of this bony destruction on imaging is critical for the diagnosis, and histopathology demonstrates similar findings to GLA in addition to increased osteoclast activity [1–3]. CCLA is caused by dysfunction of or improper drainage into the thoracic duct and retrograde reflux of lymphatic fluid with leakage resulting in pleural or pericardial effusions, ascites, protein-losing enteropathy, or generalized edema. Lymphatic imaging is typically necessary to make this diagnosis [1–4].
Increased availability of somatic genetic testing has led to the identification of multiple pathogenic variants as causes of these disorders, though an unclear genotype–phenotype correlation remains [4, 6–15]. Identification of the genotype may help to classify these diagnoses more accurately and offer insight into optimal treatments. Therefore, further understanding of the molecular etiology of these diseases is critical.
The somatic activating variant BRAF c.1799T>A p.Val600Glu (V600E) was recently described as a novel cause of macrocystic head and neck LM in three individuals [16]. This variant had been previously reported to cause other vascular anomalies, but this was the first association of this gene with lymphatic anomalies [17–20]. Since then, this variant has been attributed to three cases of CCLA in publications by our institution [15, 21]. Here, we expand upon these studies to describe six cases of GLA, KLA, and/or CCLA associated with the BRAF variant, including the three individuals previously reported as well as three new individuals. These results expand the phenotypic description of this BRAF variant beyond isolated LMs to include CLAs.
Methods
2 |
We conducted a single-center retrospective chart review at the Children’s Hospital of Philadelphia (CHOP) with all the data obtained as of February 2025. Inclusion criteria were patients with a confirmed BRAF V600E variant and a diagnosis of CLA. We queried the patient database at our institution for all patients with CLA and included those with a BRAF V600E variant. We have received an IRB exemption for this study. Genetic testing included the University of Pennsylvania Genetic Diagnostic Laboratory Somatic Overgrowth and Vascular Malformation (SOVM) Panel, CHOP’s Division of Genomic Diagnostics Solid Tumor Panel, and CHOP’s Center for Applied Genomics (CAG) research genetic panel [20].
Case Descriptions
3 |
Patient #1 was diagnosed with an isolated LM of the mesentery and gluteal region on computed tomography (CT) imaging as a neonate (Figure 1A) and underwent resection of a large portion of the mesenteric mass and partial resection of the sigmoid colon at 11 days old, followed by colostomy closure at 4 months. She then had minimal follow-up until 13 years later, when she was incidentally found to have a left-sided chylothorax on scoliosis imaging. Dynamic contrast-enhanced magnetic resonance lymphangiography (DCMRL) demonstrated disconnected inguinal and central lymphatic system and dilated thoracic duct draining to collateral lymphatic network, extending through subcutaneous tissue and left paravertebral posterior intercostal lymphatic networks (Figure 1B,C). She underwent glue embolization of left-sided paravertebral and posterior intercostal lymphatic networks, and required bilateral lymphovenous anastomosis of the inguinal skin 1 year later due to weeping lymphatic blebs in the gluteal region. Pathology from a colon resection shows dilated submucosal lymphatic channels (Figure 4A,B) and irregular mesentery lymphatics. A biopsy of the gluteal skin shows mildly dilated and irregularly shaped lymphatics in the deep dermis (Figure 4C). Lymphatics were positive for D2-40 and PROX1. At this point, overall findings were consistent with a diagnosis of CCLA. Genetic testing from the biopsy also demonstrated a somatic BRAF V600E variant at 2.2%–2.6% variant allele frequency (VAF). She was treated with sirolimus for 3 years with no clinical improvement, then transitioned to trametinib for 5 months until dermatologic toxicities, mainly persistent acne, became severe with little improvement in her condition. She was then started on a BRAF inhibitor, vemurafenib, but discontinued after 2 months due to several toxicities, including nausea, weight loss, muscle aches, and hand contractures, coupled with a lack of improvement. Though she continues to have chronic pain and discomfort, her pleural effusion has resolved, and she is able to participate in activities of daily living despite no medical treatment for 2 years.
Patient #2, with a history of left inguinal hernia repair as a toddler, was incidentally diagnosed with “lymphangiomatosis” involving the mediastinum, spleen, and mesentery on magnetic resonance imaging (MRI) (Figure 1D,E) at age 3 as part of a workup for fever without a source. She was largely asymptomatic at the time and subsequently followed for 3 years before being lost to follow-up. At age 15, she presented with bacterial pneumonia complicated by bilateral bloody chylothorax. CT imaging showed a large left pleural effusion, and subsequent MRI confirmed extensive macrocystic and microcystic lymphatic malformations in the chest, abdomen, and pelvis with splenic involvement, perivascular thickening of central vessels, and confluent marrow replacement in the vertebral bodies (Figure 1F). She had normal coagulation studies, but a markedly elevated angiopoietin-2 level of 11,759 pg/mL in blood (reference range: 1434–4141 pg/mL). Research genetic testing of cell-free DNA (cfDNA) isolated from pleural lymphatic fluid revealed a somatic BRAF V600E variant at 0.2%–2.2% VAF [20]. Magnetic resonance lymphangiography (MRL) was performed after the genetic finding revealed abnormal lymphatic drainage into the mediastinum, chest, abdomen, and pelvis (Figure 1G,H). Additionally, biopsy of the right pleura showed irregular, dilated, thin-walled vascular spaces lined by CD31^+^ , D2-40^+^ , and PROX1^+^ endothelial cells, altogether consistent with lymphatics (Figure 4D–F). Features from radiology, clinical presentation, pathology, and genetics led to a diagnosis of KLA with central conduction abnormality. She was initially treated with sirolimus, changed to selumetinib once the BRAF variant was found, and then treated with dual therapy with sirolimus and selumetinib. However, she continued to have recurrent worsening of her chylothoraces requiring frequent thoracentesis and hospitalization. As such, she recently transitioned to dual therapy with dabrafenib and trametinib and underwent lung decortication and selective glue embolization. She has demonstrated substantial clinical improvement since this surgery and the dual therapy with dabrafenib and trametinib.
At 16 months, Patient #3 was incidentally found to have a well-circumscribed mass in the posterior mediastinum and numerous splenic lesions on CT imaging of the chest obtained after recurrent acute respiratory infections (Figure 2A,B). Biopsy of the chest mass demonstrated a combined venous (CD34^+^, D240^−^, PROX1 variable) and lymphatic (D240^+^) components with focal papillary endothelial proliferation. He developed chylothorax after the biopsy for which he required thoracentesis with chest tube placement; the effusion gradually improved with dietary modification and octreotide, but did not fully resolve. At 3 years old, he underwent DCMRL, which showed hepatopulmonary perfusion leaking into the left pleural space and a dilated and tortuous thoracic duct connecting the veins and mediastinal lymphatic malformation (Figure 2D), along with selective glue embolization. Overall findings were consistent with the diagnosis of GLA with central conduction abnormality. Research genetic testing of cfDNA isolated from thoracic duct lymphatic fluid and posterior mediastinal mass biopsy identified a somatic BRAF V600E variant at 0.4% and 1.3% VAF, respectively. He responded well to diuretics and the embolization procedure and did not require targeted pharmacotherapies. The patient has remained healthy without evidence of recurrent chylothorax or lymphatic conduction abnormality for nearly 3 years.
Patient #4 was diagnosed with “lymphangiomatosis” in the mesentery and spleen on MRI (Figure 2E) at age 3, at which time she underwent excision of an abdominal LM and splenectomy. Pathology from the LM showed abnormal thin-walled lymphatic channels filled with proteinaceous fluid and irregular mural smooth muscle that showed patchy D2-40 positivity with diffuse PROX1 and CD31 staining (Figure 4G–I). She remained in good health until age 16, when she presented with protein-losing enteropathy (PLE). Subsequent MRI showed cystic lesions in the chest and retroperitoneum as well as pleural effusions. DCMRL demonstrated cystic abdominal collections with intercostal and duodenal lymphatic perfusion (Figure 2F–H). Overall findings were consistent with a diagnosis of GLA with central conduction abnormality. Genetic testing of the abdominal tissue excised 13 years prior demonstrated a somatic BRAF V600E variant at 8% VAF. She responded well to diuretics and a low-fat, high-protein diet and did not require targeted pharmacotherapies.
Patient #5 had a prenatal ultrasound suspicious for intestinal atresia, but MRI at birth demonstrated diffuse LM in the neck and abdomen, consistent with GLA. She was started on sirolimus at 2 months of age and initially did well on this medication until she developed PLE and anasarca at 16 months of age. DCMRL demonstrated mesenteric LM with bowel wall involvement as well as impaired communication between the mesenteric and central lymphatic systems (Figure 3A–D). At this point, the diagnosis evolved to that of GLA with central conduction abnormality. Genetic testing of cfDNA from pleural lymphatic fluid demonstrated a somatic BRAF V600E variant at 0.2% VAF. She has responded well to treatment with sirolimus and a low-fat diet.
Patient #6 was prenatally diagnosed with hydrops and small bowel obstruction and born prematurely at 29 weeks of gestational age. She underwent exploratory laparotomy with repair of gastric perforation on day of life 5. MRI demonstrated large volume ascites and diffuse LM involving the chest and inguinal areas, and DCMRL showed no filling of the duct after hepatic, mesenteric, or inguinal injections, indicative of primary lymphatic flow disorder (Figure 3E–I). Overall findings were consistent with GLA with central conduction abnormality. Research genetic testing of cfDNA from plasma and thoracic duct lymphatic fluid demonstrated a somatic BRAF V600E variant at 0.5% and 2.0% VAF, respectively. She is followed at an outside institution, with no further data available for review.
Discussion
4 |
This report describes six cases of CLAs associated with the pathogenic BRAF V600E variant in the absence of other known pathogenic variants. Though all six individuals had diffuse LMs and abnormal lymphatic conduction, the cases demonstrate the phenotypic heterogeneity associated with this variant and further illustrate the spectrum of morbidity associated with these disorders. To our knowledge, this is the first detailed association of the BRAF V600E with CLAs and expands the phenotypic description of this variant in patients with vascular anomalies [16–20].
The individuals described in this study underwent varied treatments, ranging from observation without intervention to dietary modification to targeted pharmacotherapies to surgical and procedural interventions. This study did not evaluate the efficacy of specific treatments in this patient population. However, our experience suggests that elucidating the genetic basis for CLAs may inform a more directed approach to treatment, especially as it relates to targeted pharmacotherapy, though additional studies are needed.
Interestingly, five of the six individuals reported in our case series developed symptoms related to abnormal lymphatic conduction only after surgical interventions. Additionally, in the majority of cases, the manifestation of this abnormal lymphatic conduction was found years after their initial diagnoses and surgeries. We are limited in our ability to determine whether these interventions may have disrupted the underlying lymphatic anatomy and caused or contributed to this abnormal conduction, or if the abnormal lymphatic conduction was an inherent aspect of these individuals’ CLA phenotypes that would develop over time. Regardless, we propose added caution when considering procedural or surgical interventions in patients with CLAs, especially in the setting of a known causative BRAF V600E variant. We also emphasize the importance of regular longitudinal follow-up even for those individuals who are seemingly asymptomatic without treatment.
CLAs are a group of diseases that can be challenging to diagnose given significant overlap in clinical presentation, imaging, and histopathology. As we develop further understanding of the molecular etiology of these disorders, it is apparent that there may be significant genetic overlap as well. Identification of the causative gene variant in an individual diagnosed with a CLA may be critical to optimizing that individual’s unique treatment plan.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Adams D and Ricci K, “Vascular Anomalies: Diagnosis of Complicated Anomalies and New Medical Treatment Options,” Hematology Oncology Clinics of North America 33, no. 3 (2019): 455–470.10.1016/j.hoc.2019.01.01131030813 · doi ↗ · pubmed ↗
- 2Iacobas I, Adams DM, Pimpalwar S, , “Multidisciplinary Guidelines for Initial Evaluation of Complicated Lymphatic Anomalies-Expert Opinion Consensus,” Pediatric Blood & Cancer 67, no. 1 (2020): e 28036.31617676 10.1002/pbc.28036 · doi ↗ · pubmed ↗
- 3Ricci K and Iacobas I, “How We Approach the Diagnosis and Management of Complex Lymphatic Anomalies,” Pediatric Blood & Cancer 69, no. Suppl 3 (2022): e 28985.33844431 10.1002/pbc.28985 · doi ↗ · pubmed ↗
- 4Grenier JM, Borst AJ, Sheppard SE, , “Pathogenic Variants in PIK 3CA Are Associated With Clinical Phenotypes of Kaposiform Lymphangiomatosis, Generalized Lymphatic Anomaly, and Central Conducting Lymphatic Anomaly,” Pediatric Blood & Cancer 70, no. 9 (2023): e 30419.10.1002/pbc.30419 PMC 1134026537194624 · doi ↗ · pubmed ↗
- 5Engel E, Le Cras T, and Ricci K, “How We Use Angiopoietin-2 in the Diagnosis and Management of Vascular Anomalies,” Pediatric Blood & Cancer 71, no. 5 (2024): e 30921.38439088 10.1002/pbc.30921 PMC 12919156 · doi ↗ · pubmed ↗
- 6Li D, Wenger TL, Seiler C, , “Pathogenic Variant in EPHB 4 Results in Central Conducting Lymphatic Anomaly,” Human Molecular Genetics 27, no. 18 (2018): 3233–3245.29905864 10.1093/hmg/ddy 218PMC 7190898 · doi ↗ · pubmed ↗
- 7Barclay SF, Inman KW, Luks VL, , “A Somatic Activating NRAS Variant Associated With Kaposiform Lymphangiomatosis,” Genetics in Medicine 21, no. 7 (2019): 1517–1524.30542204 10.1038/s 41436-018-0390-0PMC 6565516 · doi ↗ · pubmed ↗
- 8Boscolo E, Pastura P, Glaser K, , “Signaling Pathways and Inhibitors of Cells From Patients With Kaposiform Lymphangiomatosis,” Pediatric Blood & Cancer 66, no. 8 (2019): e 27790.31045327 10.1002/pbc.27790 PMC 6588438 · doi ↗ · pubmed ↗