Cationic and Neutral Heterometallic Ir-Group 12 Element Polyhydride Compounds: Synthesis, Structure and Reactivity
Amber M. Walsh, Carlos Martín-Fernández, John P. Lowe, Stuart A. Macgregor, Mary F. Mahon, Michael K. Whittlesey

TL;DR
This paper describes the synthesis and properties of new iridium-based metal complexes with hydride ligands and group 12 elements.
Contribution
The study introduces novel heterometallic Ir-group 12 element polyhydride compounds and their reactivity.
Findings
New Ir–Zn and Ir–Cd hydride complexes were synthesized through dehydrogenation and R–H elimination.
The complexes exhibit unique structures and reactivity under hydrogen exposure and Lewis base interactions.
Crystallographic and computational analyses reveal diverse hydride ligand characteristics influenced by coordination environments.
Abstract
The preparation and reactivity of some Ir–Zn and Ir–Cd heterometallic hydride complexes are described. Treatment of [Ir(IPr)2H2][BArF 4] (1; IPr = 1,3-bis(2,6-diisopropylphenyl)imidazol-2-ylidene; ArF = 3,5-C6H3(CF3)2) with M′R2 (M′ = Zn; R = Ph, Me, Et; M′ = Cd, R = Me) and Me3SiCH=CH2 results in dehydrogenation of an IPr isopropyl substituent, along with R–H elimination, to form square-pyramidal [Ir(IPr)(IPr″)(M′R)H][BArF 4] (M′R = ZnPh (4a); ZnMe (4b); ZnEt (4c); CdMe (8); IPr″ = dehydrogenated IPr) featuring apical M′R ligands. Heating 1 with 2 equiv ZnPh2 under H2 forms [Ir(IPr)2(ZnPh)2H4][BArF 4] (5) featuring trans ZnPh ligands. Exposure of 4b-c and 8 to H2 yields [Ir(IPr)(IPr″)(M′R)H3][BArF 4] (9b-c, 10) as intermediates to highly fluxional [Ir(IPr)2(M′R)(η2-H2)H3][BArF 4] (11b-c, 12). Reacting 11b–c with Lewis bases (L) effects [ZnR]+ abstraction to give…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
 1
1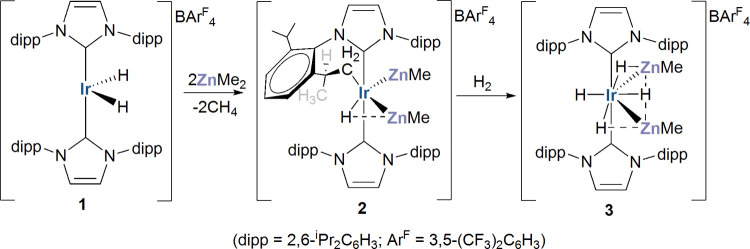 2
2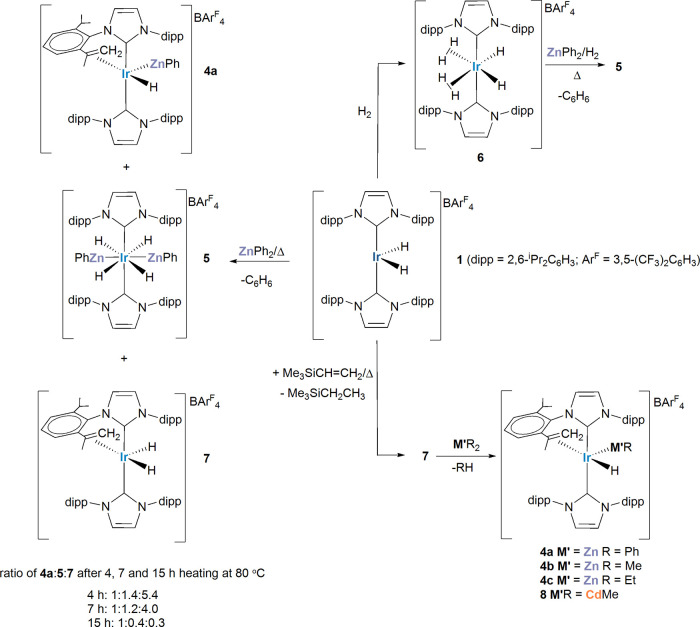 3
3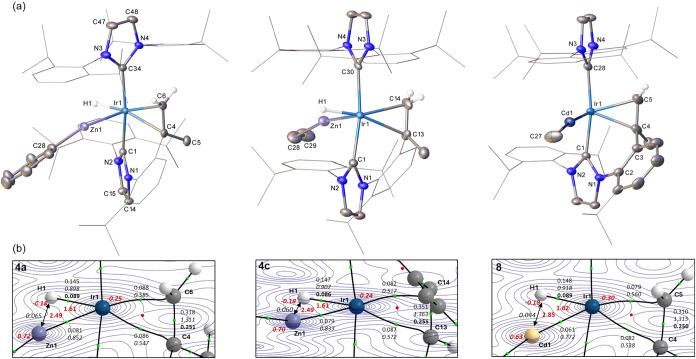 1
1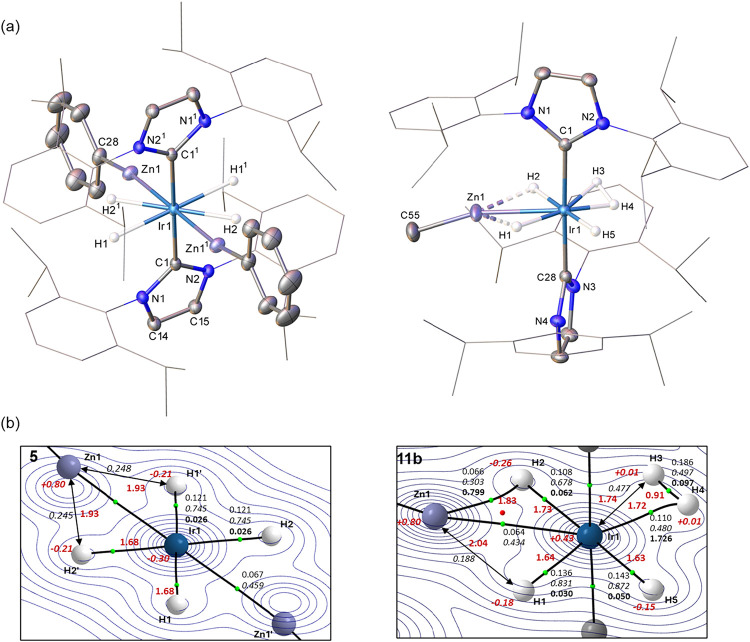 2
2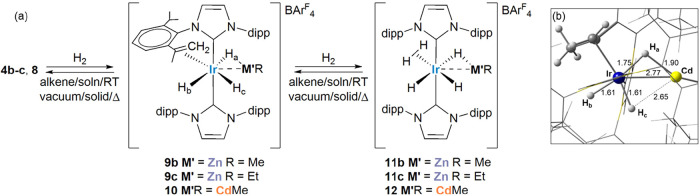 4
4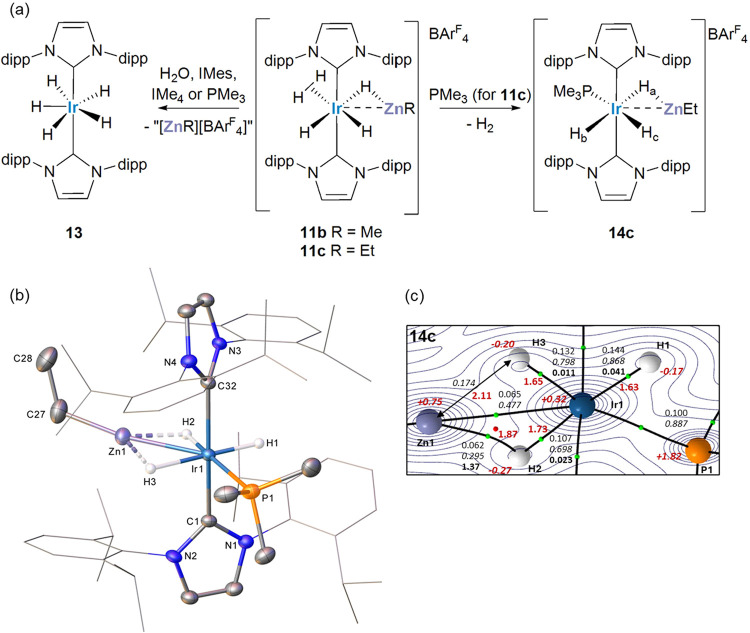 5
5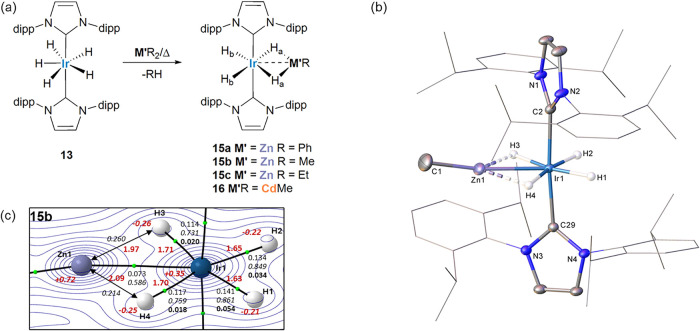 6
6- —Leverhulme Trust10.13039/501100000275
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsOrganometallic Complex Synthesis and Catalysis · Synthesis and characterization of novel inorganic/organometallic compounds · Asymmetric Hydrogenation and Catalysis
Introduction
Polyhydride transition metal (TM) compounds containing traditional Lewis-basic ancillary ligands such as phosphines and carbonyls are well-established. ?,? Until recently, examples of such TM–H_ x _ (x > 2) species featuring Lewis-acidic (LA) ligands based on s- and p-block metals were known, although not that common. ?−? ? ? ? ? ? This situation has changed over the past few years due to the renaissance of interest in TM-LA heterometallic chemistry. ?,?
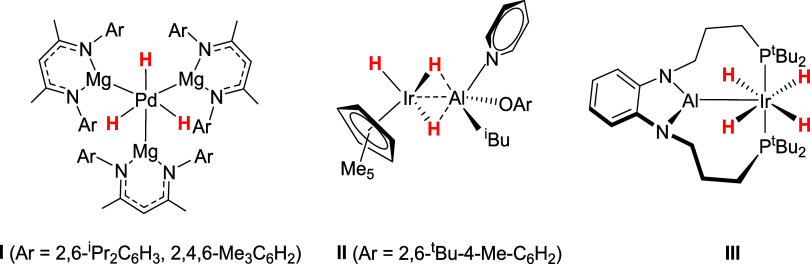
Scheme shows some recent examples which are notable for their unusual bonding interactions (I),? unexpected stoichiometric reactivity (II)? and catalytic activity with traditionally challenging substrates (III)? respectively.
Recent Examples of TM-LA Polyhydride Complexes Notable for Their Structure (I), Stoichiometric Reactivity (II) and Catalysis (III)
We recently reported? the synthesis of [Ir(IPr)2(ZnMe)2_H_4][BAr^F^ 4] (3; IPr = 1,3-bis(2,6-diisopropylphenyl)imidazol-2-ylidene; Ar^F^ = 3,5-C_6_H_3_(CF_3_)2), which was identified as a ′classical′ tetrahydride species (Scheme) on the basis of the hydride T 1 values and the splitting of the IPr carbenic carbon resonance into a ^2^ J CH-coupled quintet in the ^13^C{selective-^1^H} NMR spectrum. NMR spectroscopy also revealed that all four hydride ligands in 3 were in exchange, even at −50 °C. In stark contrast, intermolecular H/D exchange of 3 with D_2_ required heating at 40 °C, while elimination of H_2_ to afford the dihydride species [Ir(IPr)2(ZnMe)2_H_2][BAr^F^ 4] proceeded at 60 °C, but only upon heating solid 3 under constant vacuum for at least a week. Computational studies identified the important role of electrostatic H^δ−^···Zn^δ+^Me interactions in helping to stabilize 3.
Synthesis of the Heterometallic Tetrahydride Species 3
As shown in Scheme, the precursor to 3 is the cyclometalated carbene monohydride salt [Ir(IPr)(IPr′)(ZnMe)2_H][BAr^F^ 4] (2), which itself is formed in an alkane elimination reaction following the addition of 2 equiv of ZnMe_2 to the dihydride precursor 1.? Herein, we show that subjecting the latter to not only ZnMe_2_, but also ZnEt_2_ and ZnPh_2_, in the presence of an alkene affords a series of dehydrogenated carbene monohydride [Ir(IPr)(IPr″)(ZnR)H][BAr^F^ 4] products (where IPr″ has one dehydrogenated ^i^Pr substituent) which act as precursors to an array of new cationic, as well as neutral, heterometallic Ir–ZnR polyhydride species. Throughout, R–H elimination proves an efficient strategy? for the preparation of Ir–ZnR heterometallic species. As in our previous study,? CdMe_2_ shows analogous reactivity to that seen with ZnMe_2_.
Results and Discussion
Reactivity of 1 with ZnPh2: Formation of [Ir(IPr)(IPr″)(ZnPh)H][BArF
4] (4a), [Ir(IPr)2(ZnPh)2H4][BArF 4] (5) and [Ir(IPr)(IPr″)H2][BArF 4] (7)
In contrast to the near instantaneous room temperature reaction of 1 and ZnMe_2_, the reaction with ZnPh_2_ (2 equiv, C_6_H_5_F) proceeded only upon heating (80 °C) to give (over ca. 4 h) a ca. 1:1.4:5.4 ratio of three hydride-containing products, [Ir(IPr)(IPr″)(ZnPh)H][BAr^F^ 4] (4a), [Ir(IPr)2(ZnPh)2_H_4][BAr^F^ 4] (5) and [Ir(IPr)(IPr″)H_2_][BAr^F^ 4] (7). This ratio changed with additional heating (Scheme), leaving 4a as the major species after 15 h.
Reactivity of [Ir(IPr)2H2][BArF 4] (1) with ZnPh2 and Separate Syntheses of the Resulting Products and Their Derivatives
The three products were identified following independent syntheses. The nonzinc-containing dihydride salt 7 was reported previously as the main product formed upon stirring Ir(IPr)_2_H_2_Cl with Na[BAr^F^ 4] in C_6_H_5_F for ca. 12 h at room temperature; shorter reaction times (3–4 h) gave 1.? However, in our hands, 1 was the only observable species even after stirring Ir(IPr)2_H_2_Cl and Na[BAr^F^ 4] for 24 h at room temperature, or upon heating for 12 h at 60 °C. Instead, we prepared 7 in 73% isolated yield by heating 1 at 80 °C with a hydrogen acceptor, Me_3_SiCHCH_2. By ^1^H NMR spectroscopy, 7 showed two Ir–H resonances (Figure S40) at δ −13.6 and δ −41.2 (d, ^2^ J HH = 8.2 Hz) in a 1:1 ratio, the very low frequency resonance reflecting the presence of an apical hydride ligand in the structurally determined square-pyramidal structure. ?,? We are unable to explain why our data differ to those reported, a singlet of integral 2 at δ −34.0. ?,?,?
Clean formation of the Ir–ZnPh salt [Ir(IPr)(IPr″)(ZnPh)H][BAr^F^ 4] (4a) was achieved upon (i) heating 7 with ZnPh_2_ or (ii) heating 1 with a mixture of ZnPh_2_ and Me_3_SiCHCH_2_. It was identified by the presence of three ^1^H NMR resonances (δ 4.01, 3.18 and 1.48) for the dehydrogenated isopropyl substituent of the IPr″ ligand in a 1:1:3:1 ratio with a single hydride resonance at δ −7.41 (Figure S3). The X-ray structure and computational analysis of 4a (and other species in Scheme that have been characterized crystallographically) are discussed in a later section (Figures and ?).
(a) Structures of the cations in [Ir(IPr)(IPr″)(ZnPh)H][BArF 4] (4a), [Ir(IPr)(IPr″)(ZnEt)H][BArF 4] (4c), and [Ir(IPr)(IPr″)(CdMe)H][BArF 4] (8). In all cases, ellipsoids are displayed at 30% probability and nondehydrogenated IPr substituents in wireframe. Nonhydride and -alkene H atoms, as well as the minor disordered atoms in 4c and 8, are omitted for clarity. The hydride ligand in 8 was not located. (b) QTAIM molecular graphs (optimized H atom positions) with density contours in the {M′Ir1H1} plane (M′ = Zn, 4a, 4c; M′ = Cd, 8), showing computed Ir–H and Zn–H distances (Å, in red, plain text) and QTAIM atomic charges (in red, italics). BCPs (green spheres) show the associated ρ(r) (au) in plain text, delocalization indices in italics and, for bond paths to hydrogens, ellipticities in bold. M′···H1 delocalization indices also indicated.
(a) Structures of the cations in [Ir(IPr)2(ZnPh)2H4][BArF 4] (5) and [Ir(IPr)2(ZnMe)H5][BArF 4] (11b). Ellipsoids are depicted at 30% probability in both cases. Carbene substituents have been depicted as wireframes for visual ease. In 5, symmetry operation for atoms with superscripted labels: 1 1/2 – x, 3/2 – y, 1 – z; in 11b non-IPr H atoms and minor disordered atoms are omitted for clarity. (b) QTAIM molecular graphs (optimized H atom positions) with density contours in the {Zn1Ir1H1} plane showing computed Ir–H and Zn–H distances (Å, in red, plain text) and QTAIM atomic charges (in red, italics). BCPs (green spheres) show the associated ρ(r) (au) in plain text, delocalization indices in italics and, for bond paths to hydrogens, ellipticities in bold. Zn···H1 delocalization indices also indicated.
The formation of the third of the reaction products shown in Scheme, the dizinc tetrahydride salt [Ir(IPr)2(ZnPh)2_H_4][BAr^F^ 4] (5), can be attributed to the reaction of [Ir(IPr)2(η^2^-H_2_)2_H_2][BAr^F^ 4] (6), generated by addition to 1 of the H_2_ released in the formation of 4a and 7,? with 2 equiv ZnPh_2_, followed by the elimination of two molecules of benzene. In line with this, directly heating 1 at 80 °C with 2 equiv ZnPh_2_ under 1 atm H_2_ cleanly formed 5. This showed a ^1^H NMR spectrum very different (a single, temperature invariant Ir–H resonance (Figure S31) in a 4:8:24:24 ratio with the ^i^Pr methine and methyl resonances) to that of the ZnMe analogue 3 (one broad, room temperature Ir–H resonance that decoalesced into a 1:1:2 set of resonances at −50 °C), suggestive of an alternative geometry to the 1,3-arrangement of ZnR groups in 3. A 1,4-arrangement of ZnPh groups was subsequently confirmed by X-ray crystallography (Figure, vide infra).
Synthesis of [Ir(IPr)(IPr″)(M′R)H][BArF
4] (M′R = ZnMe, ZnEt, CdMe) and Reactions with H2
The formation of mono Ir–M′R species [Ir(IPr)(IPr″)(M′R)H][BAr^F^ 4] was not limited to the ZnPh derivative 4a. Treatment of 7 with ZnMe_2_, ZnEt_2_ and CdMe_2_ (all at room temperature cf. 4a) gave [Ir(IPr)(IPr″)(ZnMe)H][BAr^F^ 4] (4b), [Ir(IPr)(IPr″)(ZnEt)H][BAr^F^ 4] (4c) and [Ir(IPr)(IPr″)(CdMe)H][BAr^F^ 4] (8) respectively (Scheme).? The products were identified in the first instance by the similarity of their Ir–H chemical shifts to that of 4a (4b: δ −9.32; 4c: δ −8.07; 8: δ −9.29; Figures S10, S21, S51)? and confirmed in the cases of 4c and 8 by X-ray crystallography (vide infra).
Exposure of THF solutions of 4b-c and 8 to 1 atm H_2_ generated mixtures of the trihydride salts [Ir(IPr)(IPr″)(M′R)H_3_][BAr^F^ 4] (9b-c, 10) and pentahydride salts [Ir(IPr)2(M′R)H_5_][BAr^F^ 4] (11b-c, 12) over minutes at room temperature, with near complete conversion to the latter observed over ca. 1 h (Scheme and Figures S64 and S68). As a result of this onward reactivity, we were unable to isolate 9b-c or 10, although spectroscopically clean samples could be prepared by addition of H_2_ to samples of 4b-c and 8, followed by rapid insertion into a precooled NMR probe at 248 K (Figures S65, S69 and S72).? As for the CdMe derivatives of 2 and 3 in Scheme, the J HCd splittings on the three hydride resonances of [Ir(IPr)(IPr″)(CdMe)H_3_][BAr^F^ 4] (10: δ −8.24 (628 Hz), −12.08 (42 Hz), −14.02 (114 Hz); Figure S74) afforded information about structure.? The resonance at ca. δ −8 with the largest coupling was assigned to the hydride bridging Ir and Cd (H_a_, Schemea). This also exhibited a doublet trans-^2^ J HH splitting of 11 Hz ?,? to H_b_, for which ^2^ J HCd = 114 Hz. The remaining hydride H_c_ showed no coupling to either H_a_ or H_b_, but an NOE interaction between H_c_ and H_b_ confirmed their presence in the same molecule. ?−? ? ? In the absence of crystallographic data, full DFT optimizations on possible structures for 10 (Schemeb) supported the presence of two hydrides, H_a_ and H_c_, on either side of the Ir–Cd vector, with H_a_ (trans to the terminal Ir–H_b_ bond), interacting more strongly with the Cd center than H_c_ (trans to the alkene; computed distances: Cd–H_a_ = 1.90 Å; Cd–H_c_ = 2.65 Å). Alternative isomers (with CdMe adjacent to the alkene moiety and a dihydrogen hydride form) were significantly higher in energy (Figure S152).
(a) Formation and Reactivity of Ir–Zn/Cd Tri- and Pentahydride Salts; (b) Detail of the Computed Structure of the Cation of 10
Generation of 9b-c and 10 as trihydride species in which the IPr″ ligand is retained fits with other Ir complexes in which preferential addition of H_2_ to the metal center rather than a dehydrogenated NHC (or, indeed, dehydrogenated phosphine) ligand is also observed. ?,?
NMR studies (Figures S75–S95) of the pentahydride salts 11b-c and 12 showed them to (i) all feature two intact IPr ligands, (ii) all be highly fluxional (single hydride resonances of relative integral 5 observed at ca. δ −8 to −9 between 323 and 228 K) and (iii) exhibit short T 1 times (11b: 32 ms (248 K, 400 MHz); 11c: 45 ms (248 K, 500 MHz); 12: 45 ms (248 K, 400 MHz)), consistent with nonclassical structures (vide infra).? Given the fluxionality and nonclassical properties, the formation of free HD in the ^1^H NMR spectrum of a sample of the ZnEt derivative 11c exposed to 1 atm D_2_ was unsurprising (Figure S96). The elimination of three molecules of H_2_ from 11c to reform 4c could be brought about at room temperature in solution by addition of styrene or in the solid-state by heating 11c at 60 °C under dynamic vacuum overnight (Figures S97 and S98).
Crystallographic and Computational Studies of 4a, 4c, 5, 8 and 11b
The X-ray crystal structures of the cations in the three Ir–MR′ salts 4a, 4c and 8 are shown in Figurea. They all display distorted square-pyramidal structures, with MR′ groups in the apical site tilted over toward the hydride ligand in the basal plane (e.g., ∠alkene centroid–Ir1–Zn1 = 110.80(5)° in 4a). As in other IPr″ species, ?,? the CC bond of the dehydrogenated arm sits almost perfectly parallel to the basal plane (e.g., C1–Ir1–alkene centroid–C6 angle of 177.6° in 4a). Salts 4a and 4c, which show similar and rather short Ir–Zn distances (2.3523(3) and 2.3651(6) Å respectively) as a result of the ZnR ligands being trans to a vacant site, provide additional examples of square-pyramidal TM species with apical ZnR ligands (TM = Ru, Rh), ?−? ? ? ? and compare with related TM–HgR (TM = Rh, Ir) ?,? compounds. Salts 4a, 4c and 8 appear to be the first to present this motif in the presence of a hydride ligand, and the apical positioning of the M′R ligands indicates a higher trans influence than hydride.
The similarities between 4a, 4c and 8 are also reflected in the DFT-calculated structures, where the H atoms are optimized around fixed heavy atom positions derived from the X-ray structures. These and the associated QTAIM molecular graphs are displayed in Figureb and show computed Ir–H distances around 1.61 Å consistent with terminal hydride character. These align with both the relatively large electron densities, ρ(r), and delocalization indices, DI, at the associated bond critical points (BCPs: ρ(r) ≈ 0.15 au; DI ≈ 0.90) although the BCP ellipticities (ε ≈ 0.09) are somewhat larger than expected.? This implies some perturbation of the electron topology likely due to the cis-M′R groups, although the long Zn1···H1 distances in 4a and 4c and the low associated DIs (<0.07) indicate minimal interaction that becomes weaker still in 8. The Ir–Zn interactions in 4a and 4c are not significantly affected by the change in substituent (ρ(r) ≈ 0.08; DI ≈ 0.84) but appear somewhat stronger than the Ir–Cd interaction in 8 (ρ(r) = 0.061; DI = 0.772). This is consistent with the elongated Ir–Cd distances of the two molecules in the unit cell of 8 (2.6499(15)/2.6497(2) Å), expected on the basis of the respective covalent radii (Zn 1.22 Å, Cd 1.44 Å). ?−? ?
The X-ray structure of the cation in 5 displayed two trans-ZnPh ligands and two pairs of cis-hydride ligands placed on either side of the Zn–Ir–Zn vector in the equatorial plane (Figurea). This 1,4-trans arrangement contrasts with the 1,3-geometry found in [Ir(IPr)2(ZnMe)2_H_4][BAr^F^ 4], (3, Scheme) where one hydride sits between the two ZnMe ligands.? This difference may be a result of the increased sterics of the phenyl substituents on Zn.? Thus, the two axial IPr ligands in 5 exhibit an eclipsed arrangement of the dipp substituents that accommodates the ZnPh groups with the Ph substituents lying perpendicular to the imidazolylidene rings. The Ir–Zn distances in 5 (2.4607(3)/2.4608(3) Å) are somewhat shorter than those in 3 (2.4878(4)/2.4942(5) Å),? while still being significantly longer than the apical Ir–Zn distances in 4a and 4c.
In the X-ray crystal structure of the ZnMe pentahydride salt 11b, two hydrides (H1 and H2) were located bridging the Ir1–Zn1 bond (2.4766(4) Å), alongside a terminal hydride ligand (H5), all three of which had credible U iso values (<0.05). H3 and H4 were tentatively assigned to an η^2^-H_2_ ligand, as the U iso values were slightly larger (0.16047 and 0.09576) and restraints were necessary for placement of H3. Further insight into the H atom positions was provided by the DFT-optimized geometries in Figureb. For 5, long Ir–H distances of 1.68 Å are computed, reflecting the trans-H–Ir–H arrangement. The computed Zn1···H1/H2 distances (1.93 Å) are much shorter than in 4a and 4c above and the associated DIs are significant (ca. 0.24) indicative of some bridging character; ?,? overall the computed metrics are very similar to the symmetrical trans-H–Ir–H unit in 3.? A degree of hydride-bridging character may also attenuate the trans influence of the ZnR ligand noted in 4a and 4c, that allows them to adopt a mutually trans arrangement in 5. This is also reflected in the reduced ρ(r) and DI values associated with the Ir–Zn BCPs and the longer Ir–Zn distances compared to the equivalent metrics in 4a and 4c.
Calculations on 11b support the [Ir(IPr)2(ZnMe)(η^2^-H_2_)H_3_]^+^ formulation proposed from the X-ray study. The Ir–H5 bond exhibits terminal character (Ir–H5 = 1.63 Å; ρ(r) = 0.143, DI = 0.872, ε = 0.050) but the trans H5–Ir–H2 unit is now unsymmetrical due to the H2–Zn1 bridging interaction that weakens the Ir–H2 bond (Ir1–H2 = 1.73 Å; ρ(r) = 0.108, DI = 0.678). In this case, a Zn1–H2 bond path is seen (Zn1–H2 = 1.83 Å; ρ(r) = 0.066, DI = 0.303) as well as a far greater ellipticity at the Zn1–H2 BCP (0.799, cf. 0.062 for Ir1–H2). The Ir1–H1 bond is similar to Ir1–H5 reflecting its location trans to a weaker trans influence η^2^-H_2_ ligand and a relatively weak H1···Zn1 interaction (H1···Zn1 = 2.04 Å; DI = 0.188). Similar patterns have been seen in [Ru(IPr)2(CO)(ZnH)H_3_]^+^ and [Ru(IPr)2(CO)(ZnEt)(L)H_2_]^+^ (L = η^2^-H_2_, vacant) ?,? that also feature an unsymmetrical {TM(H)_2_M′} motif. As noted previously,? an increase in bridging character (in 11b: H5 < H1 < H2) aligns with a larger (more negative) QTAIM charge and hence greater hydridic nature.
Lewis Base-Induced Elimination of “[M′R][BArF
4]” from [Ir(IPr)2(M′R)H5][BArF 4]: Formation of Ir(IPr)2H5
During early studies of the reactions of 4b-c with H_2_, the formation of 11b-c was commonly accompanied by small, although inconsistent, amounts of the neutral pentahydride complex Ir(IPr)2_H_5 (13), which we postulate forms through adventitious moisture-induced elimination of “[ZnR][BAr^F^ 4]” (Scheme).? In support of this, dissolution of a crystalline sample of 11c in degassed, but undried THF, gave a 14% yield of 13, whereas a yield of <1% was formed in Na/K-dried THF (Figures S100–S103).
(a) [ZnR]+ Loss and Phosphine Substitution Reactions of [Ir(IPr)2(ZnR)H5][BArF 4], 11b and 11c; (b) Plot Depicting the Structure of the Cation in [Ir(IPr)2(PMe3)(ZnEt)H3][BArF 4] (14c); (c) QTAIM Molecular Graph (Optimized H Atom Positions) of 14c
We were unable to determine the structure of the “[ZnR][BAr^F^ 4]” species eliminated in the reaction. However, when 11c was dissolved in Na/K-dried THF in the presence of 1,3-bis(2,4,6-trimethylphenyl)imidazol-2-ylidene (IMes, 2 equiv), quantitative conversion to 13 was accompanied by ^1^H NMR resonances matching those of the bis-carbene adduct, [(IMes)2_ZnEt][BAr^F^ 4] (Figure S104).? Changing IMes to IMe_4 (1,3,4,5-tetramethylimidazol-2-ylidene) or PMe_3_ similarly gave 13 (Figures S105 and S113), although we were again unable to identify the eliminated carbene/phosphine [ZnEt][BAr^F^ 4] adduct in either reaction. DFT calculations showed the formation of [L_2_ZnMe]^+^ from the cation of 11b to be exergonic for all added ligands L, and most favorable when L = IMes (ΔG = −40.7 kcal/mol; all structures fully optimized and energies including a correction for THF solvent, see Table S2). ?,?
With PMe_3_, a small amount of the phosphine substitution product [Ir(IPr)2(PMe_3_)(ZnEt)H_3_][BAr^F^ 4] (14c) was also formed (Scheme). X-ray crystallography showed this to be isostructural to 11b with PMe_3_ in place of the dihydrogen ligand. The Ir(1)–Zn(1) distance of 2.4796(9) Å suggests a similar degree of Ir···Zn interaction as in 5 and 11b above. The Ir–Zn vector is straddled by hydrides H2 and H3 which have Ir–H distances of 1.56(5) and 1.86(7) Å respectively, in line with terminal and bridging character. This trend was confirmed in the computed structure and associated QTAIM metrics in Schemec, while noting the relatively large esds on the experimental Ir–H distances. Unlike in 11c, there was no evidence for fluxionality in the three hydride resonances of 14c (δ −9.37, −11.38 and −12.01) over the temperature range 328 to 193 K. Assignment of the hydride resonances as H_a_ to H_c_ (Schemea) was done on the basis of ^2^ J HP values and ^1^H,^1^H-NOESY interactions (Figure S119).
Synthesis of 13 and Reactivity with M′R2
Formulation of 13 as Ir(IPr)2_H_5 was confirmed through an alternative synthesis involving the deprotonation of the bis(dihydrogen) dihydride salt 6 ^15^ by KN(SiMe_3_)2, which afforded the complex in 71% isolated yield. In accord with the well-known phosphine analogues Ir(PCy_3_)2_H_5 and Ir(P^i^Pr_3_)2_H_5, ?−? ? the IMes derivative exhibited properties consistent with a ′classical′ pentahydride, namely a long T 1 value (938 ms, 228 K, 400 MHz; 799 ms at 298 K) for the single hydride ?,? resonance at δ –10 (the appearance of which was unchanged between 298 and 228 K) and the splitting of the Ir–C IPr resonance into a sextet (^2^ J CH = 4 Hz) in the ^13^C{selective-^1^H} NMR spectrum (Figure S111). ?−? ? Full DFT optimizations with the BP86 functional also identified a pentagonal bipyramidal pentahydride ground state structure (see Supporting Information). ?−? ? In contrast to the many known examples of late transition metal polyhydride compounds with phosphine ligands, ?,?
13 represents one of the very few known NHC analogues. ?,?−? ? ? ?
Prompted by the range of Ir–ZnR/CdR products accessible from cationic 1, the reactivity of neutral 13 with ZnR_2_ and CdMe_2_ was also investigated (Scheme). In all cases, R–H elimination took place upon heating (50–100 °C) with a slight excess of ZnR_2_ (R = Ph, Et, Me) or CdMe_2_ to yield the neutral mono-M′R tetrahydride products Ir(IPr)2(ZnR)H_4_ (15a-c) and Ir(IPr)2(CdMe)H_4_ (16). These were isolated in good yields (50–60%) as colorless microcrystalline solids that were fully characterized by 1- and 2-D NMR methods (Figures S123–S148), as well as by X-ray crystallography in the case of the ZnMe derivative 15b.
(a) Reaction of Ir(IPr)2H5 (13) with ZnR2 (R = Ph, Me, Et) and CdMe2 to form Ir(IPr)2(M′R)H4 (15a-c, 16); (b) Molecular Structure of Ir(IPr)2(ZnMe)H4 (15b); (c) QTAIM Molecular Graph (Optimized H Atom Positions) of 15b
At room temperature, the ^1^H NMR spectra of the four compounds (Figures S123, S129, S134 and S141) all exhibited two hydride resonances (1:1 relative ratio) between ca. δ −10.5 and −11.5 with T 1 times ranging from 340 to 488 ms (228 K, 400 MHz). In all cases, ROESY measurements showed the hydrides to be in exchange either at or just below room temperature (Figures S128, S133, S140 and S148); variable temperature NMR measurements on 15b showed retention of two separate resonances at all temperatures across the range 318–228 K (Figure S130). From the J HCd couplings of 350 and 95 Hz in 16, the hydrides were assigned to those bridging between Ir and Cd (H_a_, Schemea) and those terminally bound to Ir (H_b_). ?,? The IR spectra (Figure S149) were consistent with this arrangement, showing a very weak feature at ca. 2100 cm^–1^ consistent with Ir–H_terminal_ and a stronger band at ca. 1700 cm^–1^ assigned to Ir–H_bridging_.
The structure of 15b determined from the X-ray data (Schemeb) shows some asymmetry in the Ir1–H3 and Ir1–H4 distances (1.85(5) and 1.59(5) Å) and, to a lesser extent, the Ir1–H3···Zn1 and Ir1–H4···Zn1 angles (78.14°, 82.37°). The Ir···Zn distance was 2.4183(5) Å. The presence of two distinct pairs of hydrides is confirmed computationally with two shorter (terminal) Ir–H1 and Ir–H2 bonds (average data: Ir–H = 1.64 Å, ρ(r) = 0.137 au; DI = 0.855, ε = 0.044) opposite to longer Ir1–H3/Ir1–H4 bonds (average data: Ir–H = 1.70 Å, ρ(r) = 0.116 au; DI = 0.745, ε = 0.019). While no Zn1–H3/H4 bond paths are seen, the computed Zn1–H3/H4 distances (1.97/2.09 Å) and DIs (0.260/0.214) indicate some interaction is present and this is larger for H3.
Conclusions
In conclusion, we have shown that dehydrogenation of [Ir(IPr)2_H_2][BAr^F^ 4] (1) opens up further avenues for M′R_2_ species (M′= Zn; R = Ph, Me, Et; M′ = Cd, R = Me) to react with cationic Ir–H species via R–H elimination. The resulting products are the square-pyramidal salts [Ir(IPr)(IPr″)(M′R)H] [BAr^F^ 4] (4a–c, 8) which feature a dehydrogenated IPr″ ligand and axial M′R groups, showcasing the high trans influence of these Z-type ligands. Compounds 4b–c and 8 all add three molecules of H_2_ to form the highly fluxional dihydrogen trihydride salts [Ir(IPr)2(M′R)(η^2^-H_2_)H_3_][BAr^F^ 4] (11b-c, 12). The presence of water or addition of other Lewis bases induces loss of the [M′R]^+^ moiety and formation of the classical pentahydride complex Ir(IPr)2_H_5 (13), which also reacts with M′R_2_ (again via R–H elimination) to generate the complementary series of neutral heterometallic complexes, Ir(IPr)2(M′R)H_4_ (15a-c, 16). The elimination of [M′R]^+^ from 11b-c and 12 adds to our previous observations of [ZnR]^+^ and ZnR_2_ loss from cationic and neutral ruthenium–zinc heterometallic species, respectively. ?,? The breadth of such degradation reactions in transition metal-Lewis acid heterometallic chemistry is worthy of further investigation given the obvious detrimental influence it could have on catalytic applications.?
Reaction of 1 with ZnPh_2_ and H_2_ produces [Ir(IPr)2(ZnPh)2_H_4][BAr^F^ 4], 5, which exhibits a trans-1,4-arrangement of ZnPh groups. This contrasts with the 1,3-arrangement of ZnMe ligands in [Ir(IPr)2(ZnMe)2_H_4][BAr^F^ 4], that we speculate reflects the greater steric impact of the ZnPh ligand. This may also account for the somewhat more forcing conditions required for the reactions of ZnPh_2_. QTAIM analyses of the crystallographically characterized species highlight subtle trends in the {L_n_Ir(H)_2_M′R} moiety, where two hydrides straddle the Ir···Zn vector. Higher trans influence ligands, L, tend to weaken the Ir–H bond and so promote bridging interactions with the M′R unit; these patterns confirm and add to similar trends reported in earlier work on related TM–Zn systems. ?,?
Experimental Section
For general information, experimental procedures, characterization data and computational details, see the Supporting Information. Caution! Dimethyl cadmium is extremely hazardous and should be handled with utmost care using appropriate PPE. To minimize the hazards, reactions with CdMe_2_ were typically conducted on NMR tube scales using a maximum of 10 μL of a 2.4 M toluene solution of the reagent. Otherwise, no uncommon hazards are noted.
Supplementary Material
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Hlatky G. G.Crabtree R. H.Transition-metal polyhydride complexes Coord. Chem. Rev.19856514810.1016/0010-8545(85)85020-7 · doi ↗
- 2Esteruelas M. A.López A. M.Oliván M.Polyhydrides of platinum group metals: Nonclassical interactions and σ-bond activation reactions Chem. Rev.20161168770884710.1021/acs.chemrev.6b 0008027268136 · doi ↗ · pubmed ↗
- 3Bansemer R. L.Huffman J. C.Caulton K. G.A bimetallic vanadium(I) polyhydride J. Am. Chem. Soc.19831056163616410.1021/ja 00357 a 039 · doi ↗
- 4Skupiński W. A.Huffman J. C.Bruno J. W.Caulton K. G.Dinuclear elimination for rhenium hydrides and Al Me 3: Rhenium/aluminium polyhydrides J. Am. Chem. Soc.19841068128813610.1021/ja 00338 a 021 · doi ↗
- 5Barron A. R.Lyons D.Wilkinson G.Motevalli M.Howes A. J.Hursthouse M. B.Synthesis and characterization of tungsten and rhenium aluminopolyhydrides: X-ray crystal structures of (Me 3P)3H 3W(μ-H)2Al(H)(μ-O Bun)2Al(H)(μ-H)2WH 3(P Me 3)3 and (Me 3P)3H 3W(μ-H)2Al(H)(μ-H)2WH 3(P Me 3)3 J. Chem. Soc., Dalton Trans.198627928510.1039/DT 9860000279 · doi ↗
- 6Arkhireeva T. M.Bulychev B. M.Sokolova T. A.Soloveichik G. L.Belsky V. K.Boiko G. N.Reactions of tantalocene trihydride (η5-C 5H 5)2Ta H 3 with the second group metal halogenides. Crystal and molecular structure of (η5-C 5H 5)2Ta H(μ2-H)2Zn Cl 2·C 4H 8O Inorg. Chim. Acta 198814122122610.1016/S 0020-1693(00)83913-4 · doi ↗
- 7Dawson D. M.Meetsma A.Roedelof J. B.Teuben J. H.A remarkable Lewis acid-base adduct: Preparation and structure of [Ta H 5(dmpe)2][Li HB Et 3] (dmpe = 1,2-bis(dimethylphosphino)ethane)Inorg. Chim. Acta 199725923723910.1016/S 0020-1693(97)05451-0 · doi ↗
- 8Ohashi M.Matsubara K.Iizuka T.Suzuki H.Trinuclear ruthenium polyhydride complexes with a triply bridging ligand: [{(η5-C 5Me 5)Ru}3(μ3-M)(μ-H)3(μ3-H)] (M = Li, Mgi Pr, and Zn Et) and [{(η5-C 5Me 5)Ru}3(μ3-M)(μ-H)3] (M = Al Et and Ga Me)Angew. Chem., Int. Ed.20034293794010.1002/anie.20039024912596183 · doi ↗ · pubmed ↗