Guidance document on the submission of data for the evaluation of the safety and efficacy of substances for the removal of microbial surface contamination of foods of animal origin intended for human consumption
Claude Lambré, Riccardo Crebelli, Maria de Silva, Konrad Grob, Evgenia Lampi, Maria Rosaria Milana, Marja Pronk, Gilles Rivière, Mario Ščetar, Georgios Theodoridis, Els Van Hoeck, Nadia Waegeneers, Declan Bolton, Sara Bover‐Cid, Joop de Knecht, David M. Gott, Alicja Mortensen

TL;DR
The paper outlines a guidance document for evaluating the safety and effectiveness of substances used to remove microbial contamination from animal-origin foods.
Contribution
The contribution is an updated guidance framework for assessing decontaminating substances used on food.
Findings
The guidance covers chemical, biological, and combined decontaminating agents.
It emphasizes the need to evaluate safety, efficacy, resistance potential, and environmental impact.
Application methods and target pathogens must be clearly stated for proper evaluation.
Abstract
EFSA developed an updated guidance document on the submission of data for the evaluation of the safety and efficacy of decontaminating substances for the removal of microbial surface contamination of foods of animal origin intended for human consumption (referred to as ‘food’). The decontaminating substances may be either chemicals (e.g. carboxylic acids, peroxy acids or proteins), biological agents (e.g. bacteriophages) or combinations thereof. The purpose of the treatment, including the food(s) and the pathogenic microorganisms intended to be targeted, the application methods (e.g. dipping, spraying) and conditions of the treatment used (e.g. concentration) need to be stated. Information to be provided relates to (i) the safety to humans of the applied substances and their reaction products (including degradation products) remaining in the treated food; (ii) the efficacy, i.e. whether…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
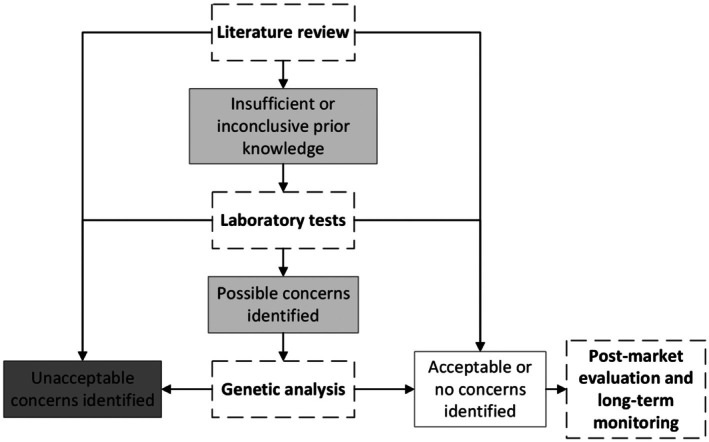 FIGURE 1
FIGURE 1Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsAgricultural safety and regulations · Antimicrobial agents and applications · Listeria monocytogenes in Food Safety
INTRODUCTION
1
EU food hygiene legislation (Regulation (EC) No 853/20041) is aimed at protecting consumers against potential risks to health and maintaining a high level of consumer protection at all stages of the food chain. This objective must be achieved by applying the appropriate measures, including good hygiene practices and hazard control measures at each step of the food chain.
According to EU scientific advice (SCVPH, 1998, 2003), decontamination practices can be a tool to further reduce the number of pathogenic microorganisms, but the use of substances intended to remove microbial surface contamination should only be permitted if a fully integrated control programme is applied throughout the entire food chain. Those substances shall be assessed thoroughly before their use is authorised.
Regulation (EC) No 853/2004 prohibits the use of any substance other than potable water (or in certain cases, clean water) to remove surface contamination from products of animal origin. However, Article 3(2) of this Regulation provides a derogation from that rule and empowers the Commission to authorise the use of substances other than potable water to remove surface contamination from products of animal origin.
Before taking any risk management decisions on their approval, a risk analysis process should be carried out, taking into account the results of a risk assessment based on the available scientific evidence and undertaken in an independent, objective and transparent manner.
In addition to the safety of the substances per se, other matters of potential concern are the potential emergence of reduced susceptibility to biocides, the potential emergence of resistance to therapeutic antimicrobials as well as the impact of the substance and its reaction products on the environment.
The present document provides guidance for dossiers on applications to be submitted to the European Commission for the authorisation of substances to be used for the removal of microbial surface contamination of foods of animal origin.
No guidance document can be exhaustive. There may be circumstances where additional data or tests to those indicated in this document are required for the evaluation. Conversely, if the applicant considers some data stipulated in the guidance as irrelevant to a particular case, these may be omitted, provided the omission is fully justified.
The impact of the treatment on product quality, on worker safety and on the acceptance of the consumer is not under the remit of this guidance document.
In this guidance document, there are references to other EFSA guidance documents which are of relevance when preparing a dossier for a decontaminating substance. Over time, these guidance documents may be updated by EFSA or new guidance documents may be developed. Therefore, when preparing a dossier in support of the safety of a decontaminating substance, the applicant should follow the most up‐to‐date version of the EFSA guidance documents cited in this guidance and of any newly available relevant guidance documents, including the Organisation for Economic Co‐operation and Development (OECD) test guidelines.
The data requirements described in this guidance document will become applicable 6 months after the date of publication of the guidance in the EFSA Journal.
Background as provided by EFSA
1.1
Through a self‐task mandate, in 2010 the BIOHAZ Panel revised the ‘Joint AFC/BIOHAZ (EFSA Panel on Food Additives, Flavourings, Processing Aids, and Food Contact Materials and EFSA Panel on Biological Hazards) guidance document on the submission of data for the evaluation of the safety and efficacy of substances for the removal of microbial surface contamination of foods of animal origin’ published in 2006 (EFSA BIOHAZ Panel, 2010). The document was intended to provide guidance for dossiers of applications to be submitted for authorisation of the substances mentioned above.
In 2020, this guidance was republished with editorial changes due to the new provisions of Regulation (EC) No 178/20022 (‘GFL Regulation’), as amended by Regulation (EU) 2019/13813 of the European Parliament and of the Council of 20 June 2019 on the transparency and sustainability of the EU risk assessment in the food chain, applicable as from 27 March 2021. The scientific content was left unchanged.
In accordance with the applicable EFSA procedures, at the beginning of the third year of the Scientific Panel mandate or at any time a need is identified (e.g. an urgent scientific need/request or annual planning), the relevant EFSA Unit places the possible review of guidance documents belonging to their remit on the agenda of the Scientific Panel plenary meeting.
At the 39th Plenary meeting of the EFSA Panel on Food Contact Materials, Enzymes and Processing aids (CEP Panel), the possible review of the sectoral guidance documents applicable in its remit was discussed. The Panel identified the need to update the above‐mentioned guidance document ‘Guidance on the evaluation of the safety and efficacy of substances for the removal of microbial surface contamination of foods of animal origin intended for human consumption’. The purpose was to specify in more detail the data and information to be provided by the applicants to EFSA.
The BIOHAZ panel was informed about this at their 166th BIOHAZ Plenary meeting and agreed that it is an opportunity to revise also certain aspects related to biological hazards, based on the lessons learned and experience obtained by the dossiers assessed so far.
Terms of Reference as provided by EFSA
1.2
The CEP Panel4 is requested by EFSA to revise the ‘Guidance on the evaluation of the safety and efficacy of substances for the removal of microbial surface contamination of foods of animal origin intended for human consumption’, taking into consideration the latest cross‐cutting EFSA guidance documents and the experience gathered over the years when dealing with these applications.
To this end, the previously published guidance (EFSA BIOHAZ Panel, 2010) should be updated, detailing thoroughly and precisely the data and information to be provided by the applicant, with regard to:
- the technical data on the substance
- the consumer exposure assessment to the substance
- the toxicological and ecotoxicological impact of the substance
- the efficacy of the substance in removing microbial surface contamination of foods
- the potential occurrence of acquired reduced susceptibility to biocides and/or resistance to therapeutic antimicrobials induced by the substance.
Public consultation
1.3
In the Plenary meetings of the FCM Panel and of the BIOHAZ Panel held on 4–5 December 2024, the draft guidance document was endorsed for public consultation on the EFSA website.
The public consultation was launched on 18 December 2024 and the comments from stakeholders were received until the 5 February 2025. The received comments and the replies given by EFSA are included in the EFSA report on the outcome of the public consultation (Annex A).
Interpretation of the Terms of References
1.4
The objective of this document is to provide guidance on the submission of data for the evaluation of the safety for consumers and environment, as well as the efficacy of substances to be used for the removal of pathogenic microorganisms from the surface of products of animal origin intended for human consumption (e.g. beef carcasses, pork meat, cheese; further referred to as ‘food’).
Although the term ‘removal’ is used in Regulation (EC) No 853/2004, in this guidance document the term ‘reduction’ was considered more applicable, because it describes both the removal (physical detachment from the surface) and the inactivation (loss of cultivability/viability) of pathogenic microorganisms. The decontamination practice is aimed at reducing the number of pathogenic microorganisms from the food surface. The achieved reduction in pathogen contamination is expected to provide benefits to public health, but the satisfactory level of public health risk reduction is a risk management decision.
The ‘decontaminating substances’ may be either chemical substances (e.g. carboxylic acids, peroxy acids or proteins), biological agents (e.g. bacteriophages), or a combination thereof. The term ‘decontamination solution’ refers to the final form of the solution as applied to the food surface, which could also contain, e.g. stabilisers and/or surface‐active agents. It could consist of either a manually prepared solution or a commercially available solution (i.e. formulated product), diluted or undiluted, or a combination of both.
With therapeutic antimicrobials are meant those antimicrobials of medical and veterinary importance for humans and animals as defined by the World Health Organization (WHO) and World Organization for Animal Health (WOAH), respectively (‘medically important antimicrobials’, WHO, 2024, 5 and future updates) and ‘veterinary critically important antimicrobial agents, veterinary highly important antimicrobial agents and veterinary important antimicrobial agents’ (WOAH, 20246 and future updates).7
The decontaminating substance should be present in the decontamination solution at the moment of application and cause a contact‐dependent inactivation during treatment or shortly thereafter (e.g. bacteriophages). As a consequence, microbial food cultures (e.g. protective or starter cultures) are out of scope of this guidance document. To the best of current knowledge, only biological agents would be bacteriophages. For products based on bacteriophages, the bacteriophage infecting the bacterial host strain and remaining in the final product is referred to as the ‘active agent’. The bacterial host strain in which the bacteriophage is replicated is referred to as the ‘production strain’.
In order to assess the safety and efficacy of the substances, the following aspects should be considered: (i) the safety to humans of the applied substances and their reaction products remaining in the treated food; (ii) the potential development of acquired reduced susceptibility to the substance or other biocides and of resistance to therapeutic antimicrobials; (iii) the efficacy, i.e. the decrease in the levels of contamination by pathogenic microorganisms; and (iv) the safety to the environment as a result of the presence of the applied substance and its reaction products in the sewage treatment plant (STP) and its effluents.
INFORMATION ON EXISTING AUTHORISATIONS AND EVALUATIONS
2
Information on existing evaluations and authorisations of the proposed decontaminating substance should be provided. This should include details of the body which carried out the evaluation and when this was undertaken. Any relevant data/studies generated/conducted in the context of other regulatory frameworks should be provided in full, including the details of the evaluation in which reference point(s) and/or health‐based guidance value(s) may have been derived.
TECHNICAL DATA
3
Identity and specifications
3.1
Chemical substance(s)
3.1.1
The identity and specifications of all substances used in the decontamination solution should be described, considering points such as:
- Composition, function and concentration of all substances in the decontamination solution, including their reaction products formed in the solution;
- Chemical names (IUPAC), CAS registry numbers, synonyms and trade names; EC numbers and REACH registration numbers;
- Molecular mass, structural formula, stereochemistry if chiral;
- Description of physico‐chemical properties: melting point, boiling point, vapour pressure, solubility in water and/or organic solvents, influence of pH on solubility, pKa (if applicable), octanol/water partition ratio, pH value of the decontamination solution;
- Source of the substance (natural vs. synthetic); if synthetic, the precursors and intermediates of the synthesis;
- Required purity of the substances used in the decontamination solution; specifications with regards to impurities8; method of analysis for the impurities;
- Particle size, shape and distribution, if applicable;
- Conditions of storage and shelf life of the substance(s) and of the decontamination solution.
For chemical substances originating from biological sources (e.g. proteins), the scientific information that enables to identify the source (e.g. genus, species, variety, strain, part of a plant source, such as roots or leaves, and organ or tissue of an animal source) should be provided, including any known toxicants that could be in the source.
Solutions from biological sources, composed of a mixture for which not all the constituents can be identified (typically plant extracts), should be characterised with regard to the constituent(s) contributing to its activity. The applicants should make reasonable efforts to fully describe the components of the mixture. For more details, the EFSA guidance on food enzymes (EFSA CEP Panel, 2021) and on feed additives (EFSA FEEDAP Panel, 2017) can be consulted. For products prepared or obtained from/with microorganisms, the EFSA guidance on the characterisation of microorganisms in support of the risk assessment of products used in the food chain (EFSA Scientific Committee, 2025a) should be consulted.
Biological agent(s)
3.1.2
As mentioned before, among biological agents used in the decontamination solution, to date, only bacteriophages can be considered. For bacteriophages, the characterisation applies to both the bacteriophage and its host strain. The characterisation of microorganisms should be generally performed by whole genome sequencing (WGS)‐based analyses. The WGS data for the strain under assessment should provide information on the taxonomic identity of the strain, as well as information on its characterisation regarding the genetic modifications, if any, and the potential presence of genes of concern. Genes of concern are those known to contribute to the production of toxins, harmful metabolites, therapeutic antimicrobials, acquired gene(s) conferring resistance to therapeutic antimicrobials, and those coding for virulence factors.
In summary, the applicant is asked to provide: (i) the origin and history of the genetic modifications, if any, as well as the taxonomic identification of the specific production strain and bacteriophage under assessment; (ii) the presence in the production strain and/or the bacteriophage of genes involved in resistance to therapeutic antimicrobials or encoding the production of therapeutic antimicrobials; (iii) information related to toxigenicity and pathogenicity (including infectivity) of the production strain as well as for its bacteriophages, the investigation of the presence of genes coding for toxins and other virulence factors, genes coding for lysogeny and genetic elements known to be involved in transduction, including the host range for bacteriophages on a representative set of strains belonging to the target and closely related bacterial species; and (iv) the presence of viable cells and DNA from the production strain in the final product, when relevant.
For this, the applicant is referred to section 3 of the EFSA guidance on the characterisation of microorganisms in support of the risk assessment of products used in the food chain (characterisation of the microorganism) (EFSA Scientific Committee, 2025a) and its subsections 3.1 (taxonomic identification), 3.2 (antimicrobial resistance), 3.3 (production of antimicrobial substances), 3.4 (toxigenicity and pathogenicity), 3.5 (genetic modifications) as well as section 4 (presence of viable cells and DNA in the final product).
Furthermore, the following should be provided for bacteriophages:
- The particle size of the bacteriophage;
- The form of the bacteriophage in the product (e.g. free, encapsulated) and, when encapsulated, the description of the structure, the carrier and the quantity of the active agent per encapsulation particle;
- Concentration of formulations (phage titres), as well as volumes applied and, absolute numbers of phages delivered;
- The purity specifications of the (bacteriophage) solution; specifications with regards to impurities and method of analysis for the impurities;
- The conditions of storage and shelf‐life of the decontamination solution, so the phage retains its activity;
- The activity of the bacteriophage at the conditions foreseen in the product, such as various temperatures (optimum and range), pH (optimum and range), water activity (optimum and range), and other factors, like NaCl concentrations, providing the results of the plaque assay and/or planktonic killing assay;
- Information on the decontamination approach (passive/active) (EFSA BIOHAZ Panel, 2009).
Manufacturing process and processing procedures
3.2
Decontaminating substances
3.2.1
To define the critical points that determine the purity and impurities of the decontaminating substance, a detailed description of the manufacturing process should be provided, such as chemical synthesis, fermentation, cultivation, extraction from organic material or distillation and downstream purification steps, supported by a flow chart, if appropriate.
For bacteriophages, the composition of the fermentation/cultivation media should be provided, as well as any chemicals used for phage purification. For genetically modified microorganisms used as production strains and grown under contained conditions, Directive 2009/41/EC9 applies.
Decontamination solution
3.2.2
The preparation of the decontamination solution should be described. The key stages, including the point(s) of introduction of the decontaminating substances and other components into the solution, as well as any subsequent process steps, should be provided, supported by a flow chart, if appropriate.
Conditions of use of the decontamination solution
3.3
The purpose of the treatment should be described, including the foods intended to be treated and, for each food, the pathogenic microorganisms that the substance is intended to target. A brief statement of the purpose of the treatment within the Food Safety Management System (FSMS) of the target foods should also be provided to justify the requirement for the treatment as a food safety intervention.
The following points should be included when describing the decontamination process:
- Point(s) in the processing lines in which the decontamination solution is intended to be applied, including any instances where the treatment may be repeated at multiple stages of processing, if applicable.
- Methods of application of the decontamination solution (e.g. dipping, spraying).
- Conditions during the application, including the volume of solution used, concentration of the active substance(s), temperature and pH of the solution and the target, duration and pressures applied. All conditions should be described with a range specifying the minimum and maximum values.
- Approximate volume of the solution per mass and surface area of treated food (possibly values at several stages).
- Subsequent removal of the decontamination solution from the food (including the conditions used), if applicable (e.g. washing or trimming of the treated area).
- Recycling of the decontamination solution or substance(s) thereof, the conditions used, if applicable.
- Amount of decontamination solution running off per time (e.g. in litres per day).
- For bacteriophages, both the duration of the application and the time needed for the agents to contact their host bacteria and kill them.
Methods of analysis
3.4
All methods should be provided that are used for the microbial analyses as well as the measurement of all substances in the decontamination solution and their reaction products that may remain in the treated food and/or in the wastewater (including protocols, validity and performance parameters).
ASSESSMENT OF SAFETY TO HUMANS
4
Chemical substance(s)
4.1
The safety assessment of decontaminating substances considered as chemicals (definition in the Interpretation of the ToR) should include both the estimation of consumer exposure (Section 4.1.1) and the toxicological assessments (Section 4.1.2).
Consumer exposure assessment
4.1.1
For the exposure assessment, needed for risk characterisation, applicants should provide the amounts or concentrations of all substances in the decontamination solution and their reaction products that may remain in the treated food (see Section 3.1). The assessment should be carried out by a dedicated tool in order to follow a harmonised approach (see Section 4.1.1.3).
Occurrence of substances in food
4.1.1.1
As a conservative approach, the amount of the substances (including their reaction products formed in the solution) remaining on the food, can be derived from the amount of decontamination solution applied per mass or surface area of treated food. To estimate the consumer exposure, these data have to be converted to a concentration expressed in mg/kg food. For a more refined estimate, the amount of solution retained on the food surface may be determined gravimetrically using small specimens with a high surface to mass ratio.
Depending on the substance, the Panel may request additional data on potential reaction products formed in the food.
Food consumption data
4.1.1.2
Since 2010, the EFSA Comprehensive Food Consumption Database (Comprehensive DB) has been regularly populated with the most recent individual‐based data on food consumption provided by competent authorities in the European countries (EFSA, 2011). Currently, food consumption data covering different population classes from infants to elderly are available from dietary surveys carried out in the majority of the EU Member States.
While the concentration data reported for decontaminating substances refer in most cases to the raw primary commodities (RPCs) (e.g. pork meat), the Comprehensive DB includes consumption data for foods as consumed, such as composite dishes (e.g. prepared meat salad) and food products (e.g. sausage). In order to match the consumption data with the available residue data for decontaminating substances, the consumption data reported in the Comprehensive DB have been disaggregated to raw primary commodity derivatives (RPCDs) (e.g. cheese, fried meat) and converted into RPC equivalents. The procedure involved identification and quantification of the single ingredients present in each composite food and a subsequent conversion of these single ingredients into their RPCs by means of conversion factors, where relevant. These data are retrieved from the EFSA RPC Consumption database, based on the principles described in details in the technical report of the RPC model (EFSA, 2019a), thus using the dietary surveys submitted to EFSA before 31st March 2018.
An overview of the surveys, countries and population classes, together with the methodology of the dietary data collection, is available in Appendix B (Table B1).
FoodEx2 System
The codes of the FoodEx2 classification system are used to match the occurrence data with the RPC consumption data for the purpose of the exposure assessments. This classification system has been deployed by EFSA across regulatory domains and successfully implemented for the annual collection of pesticide residue monitoring data. FoodEx2 describes foods and food groups with a five‐character alphanumerical base term, which can then be organised according to different hierarchies, depending on the domain of interest. In addition, facet descriptors can be used to add further descriptions of the foods consumed in relation to different properties and aspects of the foods. Details on the FoodEx2 classification system are available in the dedicated page of the EFSA website.10
Exposure assessment methodology
4.1.1.3
For the exposure assessment, the applicant should use the EFSA exposure calculating tool ‘Pesticide Residue Intake Model’, revision 4 (PRIMo 4).11 It is an online tool designed for complex assessments in the field of pesticide residues, but its core functionalities are also applicable for decontaminating substances. The tool uses individual consumption and body weight data from the RPC consumption database and ensures a harmonised methodology.
A technical report describing the use and the underlying principles of PRIMo 4 is available to facilitate its application (EFSA, 2024).
According to the agreed methodology (EFSA, 2011), for chronic exposure assessments, dietary surveys with only one reporting day per individual are not considered, as they are not adequate to assess repeated exposure. For each individual, the total relevant residues are combined with the average daily consumptions of the corresponding food commodities. The resulting exposures per food are summed in order to obtain total chronic exposure at individual level (standardised by using the individual body weight). The mean and the highest reliable percentile (usually the 95th percentile) of the individual exposures are subsequently separately calculated for each dietary survey and each population group. Results can be exported in Excel format from the PRIMo 4 tool.
The exposure estimates from the PRIMo 4 tool are expressed as mg of the substance/kg body weight (bw) per day.
An uncertainty analysis on data availability and exposure assessment should also be provided based on the recommendations of the EFSA Guidance on Uncertainty Analysis in Scientific Assessments (EFSA SC, 2018).
Toxicological assessment
4.1.2
In line with the data requirements of the present guidance document, a description of the existing toxicological data (Section 4.1.2.1), or the justification of the testing strategy chosen by the applicant to demonstrate the safety of the proposed decontaminating substance (Section 4.1.2.2), should be provided in the technical dossier. This should include the rationale for any inclusion or exclusion of specific types of in vitro*/*in vivo toxicity studies, developed in line with the tiered approach described in section 5 of the EFSA Food Additives (FAs) guidance document (EFSA FAF Panel, 2026).
The toxicological data for each component of the decontamination solution, including impurities and reaction products that may remain in the treated food, are the basis for hazard identification and hazard characterisation. The data should include dose–response relationships (full study reports). However, full hazard characterisation will not be needed for reactions products and impurities for which the exposure remains below the applicable threshold of toxicological concern (TTC) (see ‘Assessment of reaction products and impurities remaining in food’ under Section 4.1.2.2).
Assessments of the decontaminating substances and reaction products within other regulatory frameworks (e.g. ECHA, EMA, FSA or FDA) can only be utilised provided the original data are submitted to EFSA and the ownership of the data is guaranteed. If only summary data are provided, this could be considered as supportive information but not used for the risk assessment.
Substances already authorised to be used in food in the EU
4.1.2.1
For substances already approved for direct addition to food in the EU, such as food additives (Reg. EC 1333/200812), the assessment could be based on the existing toxicological data and the applicant should provide:
- Confirmation that the decontaminating substance(s) are compliant with the relevant EU specifications.
- Information on existing authorisations and evaluations, including:
- name of the institution that carried out the evaluation;
- date of the evaluation;
- summary of the evaluation, identifying the critical studies and their no‐observed‐adverse‐effect‐levels (NOAELs)/lowest observed adverse effect levels (LOAELs) or benchmark dose lower confidence limit (BMDL) values;
- health‐based guidance values (HBGVs), the uncertainty factors applied, and any other uncertainties described in the evaluation.
If there are changes in the manufacturing process of already authorised substances, their specifications or conditions of use, the applicant needs to re‐assess the safety, either by providing new data or a scientific justification for why new data are not necessary.
Substances not authorised to be used in food in the EU
4.1.2.2
For substances not authorised to be used in food in the EU, it is recommended to follow the tiered approach for conducting toxicokinetic and toxicity studies described in section 5 of the EFSA FAs guidance document (EFSA FAF Panel, 2026). It is designed to evaluate the following core areas of the safety assessment of the proposed decontaminating substance:
- – genotoxicity
- – toxicokinetics
- – toxicity other than genotoxicity, to identify toxicity in target organs following repeated dosing, reproductive and developmental toxicity and carcinogenicity.
The relevant information is obtained by in vitro and in vivo experimental studies. Data from studies in humans, when existing, are considered in the weight of evidence, together with the data from the in vivo experimental animal studies. Information should be obtained by non‐animal testing if feasible, in accordance with the requirements described in section 5.4 of the FAs Guidance document (EFSA FAF Panel, 2026) and in line with the 3Rs (replacement, reduction and refinement) principles (Directive 2010/63/EU13). The full reports of these studies and of epidemiological studies as well as their compliance with international or national guidelines or Good Laboratory Practice (GLP) should be included, together with any deviations from these.
The tiered approach for toxicokinetic and toxicity testing recommends animal testing only until the available information is sufficient to reach a conclusion in the risk assessment. It consists of four tiers, for which a description of the data requirements and the triggers prompting the need to move to a higher tier, are described in detail in sections 5.4–5.8 of the EFSA FAs Guidance document (EFSA FAF Panel, 2026). The genotoxicity assessment should be performed according to the testing strategy described in the EFSA FAs Guidance and in the cross‐cutting SC EFSA Guidance documents on Genotoxicity (EFSA, 2023; EFSA SC, 2011, 2017, 2021a), and should be performed for each individual component of the decontamination solution.
A flow chart of the tiered approach is shown in Appendix A (Figure A1).
In case the substances are not absorbed, and considering their chemical reactivity owing to their intended decontaminating activity, special attention should be paid by the applicant to potential local effects on the gastrointestinal tract following oral exposure.
This approach may permit to identify a reference point (RP; e.g. NOAEL or BMDL) and to derive a HBGV (hazard characterisation) from the RP, using appropriate uncertainty factors (see SC guidance on default values, EFSA Scientific Committee, 2012a). For risk characterisation, the evaluation of human exposure to the decontaminating substance needs to be compared with the HBGV. In case a HBGV could not be derived, a margin of exposure (MOE) approach could be considered.
Allergenicity
If a component of the decontamination solution is a potential allergen (e.g. a protein) or contains residues of proteins or other known potential allergenic molecules, the data requirements described in section 10 of the EFSA Guidance on Novel Food (EFSA NDA Panel, 2024) should be followed.
Assessment of reaction products and impurities remaining in food
For reaction products and impurities for which a RP or a HBGV is available based on relevant toxicological data, the risk assessment should use both the highest mean and 95th percentile (high consumers) exposure estimates (see section 1.7 and 4.5 of EFSA FAs guidance, EFSA FAF Panel, 2026).
If toxicological data (as described in the tiered approach) are not available, non‐testing methods may be used for a preliminary assessment of their toxicological potential, including grouping and ‘read‐across’ (see section 5.4 of the FAs Guidance), computational methods (structure–activity relationships (SAR), quantitative structure–activity relationships (QSAR)) and the TTC approach (EFSA Scientific Committee, 2019).
For reaction products and impurities that are both genotoxic and carcinogenic, the Scientific Committee states that the MOE approach can be applied (EFSA Scientific Committee, 2012b; EFSA Scientific Committee, 2025b), which means that limits of such reaction products and impurities in the specifications should be as low as reasonably achievable and should result in an MOE of at least 10,000.
For non‐genotoxic reaction products and impurities for which no toxicity data are available, the TTC approach is applied. In this case, the Panel checks if the high exposure estimates based on the most relevant scenario used for the risk assessment of the proposed decontaminating substance, using both the highest mean and 95th percentile exposure estimates, are below the relevant Cramer Class TTC values (i.e. 1800, 540, 90 μg/person per day for Cramer I, II, III substances, respectively, Cramer et al., 1978; Munro et al., 1996).
For genotoxic compounds for which no carcinogenicity data are available, the TTC approach is also applied. In this case, the Panel checks whether the high exposure estimates based on the most relevant scenario used for the risk assessment of the proposed decontaminating substance, using both the highest mean and 95th percentile exposure estimates, are below the TTC for genotoxic compounds of 0.15 μg/person per day (i.e. 0.0025 μg/kg bw per day; EFSA Scientific Committee, 2019).
Biological agent(s)
4.2
For biological agents proposed as decontaminating substances, applicants are advised to refer to the EFSA guidance on the characterisation of microorganisms in support of the risk assessment of products used in the food chain (EFSA Scientific Committee, 2025a). This guidance outlines the comprehensive requirements for the safety assessment of biological agents used within the food chain. If various formulations are foreseen, all of these should be tested and any relevant evidence available should be included.
For bacteriophage‐based decontamination solutions, specific testing and data submission requirements include the characterisation of the bacteriophage and the production strain(s), as detailed in section 3 of EFSA Scientific Committee (2025a) and summarised in Section 3.1.2 of this guidance. Additionally, applicants must demonstrate the absence of viable cells of the production strain and, in case the bacterial host strain harbours genes of concern (e.g. acquired AMR genes) or is genetically modified, they must demonstrate also the absence of DNA from the production strain in the final product (section 4 of EFSA Scientific Committee, 2025a).
The possible impact of the products under scope on the gut microbiome, should be considered in certain cases (e.g. when an adverse effect can be anticipated based on the body of knowledge, or when their use is expected to have effects on the gut microbiome of animals or humans), and will be assessed on a case‐by‐case basis. Examples of adverse effects on the gut microbiome include gut dysbiosis triggering colitis, diarrhoea or shedding of pathogenic microorganisms (EFSA Scientific Committee, 2025a).
Where the bacteriophage itself or a component thereof is a potential allergen, the applicant is referred to section 10.4 (Novel foods for which the allergenic potential is unknown) of the Guidance on Novel Food (EFSA NDA Panel, 2024).
For a biological agent, the applicant should provide a description of its reactions and fate in the treated food. This includes the quantification of residual levels of the active agent used in the treated food.
EFFICACY OF PATHOGEN REDUCTION
5
The application of decontaminating substances under defined conditions (as specified in Section 3.3) will be regarded efficacious, when a reduction of the prevalence and/or numbers of target pathogenic microorganisms is consistently achieved (i.e. considering variation between experiments, or between batches of naturally contaminated foods) and is statistically significant compared to a proper control sample. The control sample can be treated with water (e.g. in case of decontamination of meat carcasses and cuts that can be treated with water and cause a reduction of the microbial load by a physical rinsing effect) or left untreated in case the treatment does not replace a water treatment (e.g. decontamination of ready‐to‐eat food) or is not applied as a liquid with run‐off. The enumeration error is to be considered for defining whether the observed reduction is relevant, especially for log reductions below 1 log.
Relevant studies for the dossier include existing experimental work (derived from the literature) complemented by experiments performed specifically for the dossier (so called ‘in‐house studies’). The dossier intended to assess efficacy should include full reports of relevant experiments and provide at a minimum the information detailed below.
To assess the comprehensiveness of the evidence of existing experimental work, information on how the studies were retrieved should be provided. For example, the search strategy (e.g. exact search string used for each literature database, date of the searches, search limits) and the study selection process used to identify relevant studies should be thoroughly documented. Examples can be found in EFSA BIOHAZ Panel (2016) and EFSA CEP Panel (2018).
Studies conducted within the purpose of the treatment (e.g. target pathogens) and under the defined conditions of application of the substance and within the range of parameters that the decontamination solution is intended to be used are to be provided, based on the information in Section 3.3. Examples of such requirements, for both studies retrieved from the literature and for in‐house studies, include:
- the stage of application along the processing line (including the scope of repeated or combined treatments) on a specific food [population];
- the use of the specified mode of application using a concentration and temperature of the solution as well as pressure and duration of treatment within the specified range [intervention];
- the inclusion of a proper control, e.g. that is either treated with water or left untreated in case the treatment does not replace a water treatment or is not applied as a liquid with run‐off [comparator];
- the testing of at least one of the target pathogenic microorganisms or indicator microorganisms (e.g. Enterobacteriaceae counts, coliform and/or Escherichia coli) at a minimum immediately after treatment and optionally also during storage and at the end of shelf‐life [outcome of interest]; and
- whether studies are experimentally controlled undertaken in a laboratory, or in pilot‐scale plant, or in an industrial (commercial) setting [study design and setting].
The dossier should include a coherent presentation of the arguments for the use of the decontamination solution, supported by studies of the efficacy of pathogen reduction and the potential development of acquired reduced susceptibility to the formulated product itself, performed according to the guidelines below and presented in a structured way. It is recommended that each of the items below is addressed in a summary and cross‐referenced to appropriate enclosures or annexes:
- All studies should be made with the solution for which authorisation is sought, at a specified range of concentrations. If various formulations are foreseen, all of these should be tested and any relevant evidence available should be included.
- The processing conditions used to evaluate the efficacy must be comparable with those for which the formulated product is intended (e.g. using a concentration and temperature of the solution as well as duration of treatment within the specified range). Care should be taken that the substance is evenly distributed over the food sample surface. The study design should be as close as possible to the real (industrial/commercial) conditions under which the formulated product is intended to be applied, including the aforementioned option of consecutive applications of the formulation/substance within a single process. Thus, pilot or in‐plant (if feasible) studies are more appropriate than laboratory‐scale studies. Moreover, if the formulated product is intended, for example, to be used as a dip or spray on broiler carcasses with skin, then broiler carcass samples with skin should be dipped or sprayed, as appropriate, in the experimental study.
- Justification of the concentration of the product formulation proposed should be experimentally demonstrated, for instance by providing data, showing the effect of different concentrations of the product formulation on the target pathogenic microorganisms reflective of the conditions of use.
- Although the treatment is intended to reduce the target pathogenic microorganisms, data on non‐pathogenic microorganisms, such as indicator microorganisms (e.g. Enterobacteriaceae counts, coliform and/or Escherichia coli) should be provided, when relevant, as these data may assist in the assessment of the overall efficacy of the proposed application.
- The study must include a comparison of the prevalence (number of contaminated units over the total number of units) and/or numbers (concentration) of the pathogenic/indicator microorganisms on the food treated with the decontamination solution and on the food of the control group (i.e. on the food treated with water or left untreated). The selection of the proper control treatment should be justified. It is recommended that the only difference between the treated and control group is the presence or absence of the substance and not the method of application or other factors (e.g. inoculated with the target organism using the same procedure, stored at the same temperature and under the same storage conditions, same detection and/or enumeration method used). Comparisons of samples treated with water and those left untreated can be provided as supportive evidence of the rinsing (physical removal) effect of the water treatment.
- Studies involving artificial inoculation must be made with inoculated target pathogenic microorganisms, taking into account strain diversity. This can be achieved by using different strains or cocktails of strains, including standard reference strains (for comparison with other studies), strains isolated from the surface of the foods of animal origin to be treated and/or clinical strains. Emphasis should be given to the use of serotypes/genotypes of (epidemiological) relevance to the target food. Furthermore, care should be taken that the inoculum is evenly distributed over the food surface and that the time between inoculation of the target organism and treatment with the substance is sufficient to allow attachment of the bacteria, mimicking the attachment strength of microorganisms to the matrix in natural settings (e.g. at least 15 min). An inoculum should be tested at a range of levels including the level expected in the food product. The same inoculum should be used for control and treated food and it should be high enough to quantify the log reduction of the target pathogen and enable the comparison of log reductions of the target microorganisms between the two experimental treatments. Of note is that the food surface can be inoculated with laboratory prepared cultures (in suspension or dry form), or with faecal material inoculated or not with laboratory prepared cultures (in suspension or dry form).
- Sampling is to be done at key time points during testing (e.g. immediately before treatment, immediately after treatment (considering the contact time and removal of the substance, if applicable) and – optionally – during subsequent storage and at the end of shelf‐life). The same testing should also be followed for the foods used as controls, i.e. samples treated with water or left untreated.
- It is recommended to use a validated reference method or parts thereof (e.g. FDA method, ISO method) or an acceptable method other than a validated reference method (e.g. Petri film), that maximises the recovery of the bacteria, for the detection and enumeration of the target organism. Studies are recommended that include a recovery technique to allow stressed cells to recover (e.g. comparison between selective and non‐selective media, thin layer agar, prolonged incubation) as these studies are more reliable compared to those that do not measure the stressed cell population and may therefore overestimate the effectiveness of the treatment. The determination of the efficacy of a formulated product must involve the use of an appropriate neutralisation method (ISO 18593:201814) or the removal of the formulated product. When assessing the efficacy of bacteriophages in reducing bacterial contamination on food surfaces, one approach involves neutralising the residual effect of phages by centrifugation of the sampled food and removing the supernatant that contains the phage particles. The bacteria that have survived the phage treatment remain in the precipitate. They are re‐suspended and enumerated after serial dilutions and plating (Dhowlaghar & Denes, 2023; Soni et al., 2010).
- For studies in pilot‐scale plants or in industrial settings, redistribution of the target organisms on the surface, e.g. due to translocation of cells caused by liquid treatments (spraying, dipping into the decontamination solution) needs to be assessed.
- The study design must be justified in relation to the intended use of the formulated product and must include a sound statistical methodology to demonstrate the hypothesis to be tested (i.e. efficacy in relation to the control). The design, particularly in terms of the number of samples (or ‘sample size’) required to demonstrate the claimed efficacy, must be justified and take into account: (a) the magnitude of the expected effect (or ‘effect size’); (b) the ‘significance level’ (noted α), that is the probability to conclude that the substance is efficacious when it is not; (c) the ‘power’ of the study (noted 1 − 𝛽), that is probability to show that the substance is efficacious when it is truly present; and (d) the measurement variance (analytical uncertainties and sources of variability).
- It is required to perform a statistical test (e.g. ANOVA, t‐test, post hoc test) for in‐house studies and to use independent experimental trials using independent samples (replicates) per trial, to increase the statistical power of the findings about the efficacy of the decontamination solution under assessment. The enumeration error is to be considered for defining whether the reduction is relevant, especially for log reductions below 1 log.
- The factors and their levels that may influence the efficacy of the substance (e.g. organic load, pH, temperature of the solution or the food) must be provided, along with the methods used to control and monitor the concentration and temperature of the decontamination solution in the processing plant during operational time. These parameters provide the basis for setting the minimum HACCP criteria and control parameters for operational monitoring.
- The possible development of acquired reduced susceptibility to the formulated product under assessment needs to be evaluated (see Section 6).
For each experiment, it is required to state the experimental setting (laboratory scale, pilot‐scale or industrial scale), the type of contamination (naturally present or artificial contamination by inoculating the samples with the target microorganism, including the case where faeces were used as vehicles of bacterial contamination), the substance or the composition of the formulation, the application method (e.g. spraying, dipping or else), the product type/description (e.g. pork carcass pre‐chill, cheese) and the product‐specific subcategory (e.g. as described in the publication). Further, the following information needs to be stated:
- the treatment characteristics: the concentration and temperature of the decontamination solution, as well as the duration of treatment of the application; and, additionally, when available, the pH (e.g. organic acids) of the decontamination solution and the pressure of the application.
- the contamination characteristics: the bacterial group (e.g. the pathogens Salmonella spp., Listeria spp., and indicator organisms Enterobacteriaceae, coliforms, E. coli) and subgroup (when available). In case of artificial inoculation, information to be provided, when available, is the origin of the strain(s), the pool of strains (if applicable) and the inoculum preparation of the strains (including the growth phase of the culture, or the stress conditions applied during inoculum preparation, when relevant, e.g. in habituation/adaptation of the inoculum, to mimic the stress history of cells to be exposed to the decontamination solution) as well as the inoculation procedure.
- the analytical methods: the analytical method used for detection or for enumeration, the method of sampling and the limit of detection and the limit of quantification (in case of non‐quantifiable survivors). The enumeration error is to be considered for defining whether the reduction is relevant, especially for log reductions below 1 log.
- the treatment and storage conditions of food after treatment: the treatment of samples, i.e. untreated (control), water (control), decontamination solution, the timing of sampling (i.e. before treatment, immediately after treatment, during storage, end of shelf‐life) and, if storage is applied, the storage conditions (i.e. temperature, duration, other conditions, e.g. packaging).
- the outcome reporting: the number of samples tested (per sampling point and independent trials), the microbial concentration (central measure, dispersion measure and units) when enumeration was performed for samples that have been treated with water or the decontamination solution or are left untreated, the number of positive samples and number of samples tested or proportion of positive samples when presence/absence testing was performed. A series of calculations can be applied to the data to harmonise the measurement unit used in the various experiments. For the comparison and the evaluation of experiments, results are to be reported as the mean log_10_ reduction. This is the difference between the means of the log_10_ concentration of control group (treated with water or left untreated) and treated group (treated with the decontamination solution). Please refer to the EFSA CEP Panel (2018) for a description of how the mean log_10_ reductions and corresponding 95% confidence intervals of the log_10_ reductions can be calculated. Also, the statistical analysis used needs to be reported, including treatment of negative samples in quantitative analysis.
POTENTIAL EMERGENCE OF ACQUIRED REDUCED SUSCEPTIBILITY TO THE DECONTAMINATION SUBSTANCE AND/OR OTHER BIOCIDES AND/OR RESISTANCE TO THERAPEUTIC ANTIMICROBIALS
6
Information should be provided to assess whether a decontaminating substance (chemical substance or biological agent) present in the decontamination solution (or in the formulated product(s), if applicable) may promote the acquisition of reduced susceptibility to itself in strains from target and non‐target species. In the case of bacteriophages, the assessment of resistance development focuses exclusively on target species.
If acquired reduced susceptibility to the substance or formulated product is observed, further testing should assess whether its use could also promote acquisition of reduced susceptibility to other biocides and/or resistance to therapeutic antimicrobials. This can occur through ‘cross‐resistance’, where a single mechanism confers reduced susceptibility or resistance to both the substance/formulated product and other antimicrobials and/or ‘co‐resistance’ where resistance to the substance is encoded on a genetic element that also carries other distinct resistance genes. The evaluation for reduced susceptibility to other biocides should prioritise those likely to be used in the industrial settings where the decontamination solution is intended to be applied.
An analysis needs to be performed to understand how acquired reduced susceptibility to the decontaminating substance (or formulated product) could contribute to acquired reduced susceptibility to other biocides and/or resistance to therapeutic antimicrobials. Additionally, the analysis should evaluate the likelihood of bacteria to acquire multiple resistance mechanisms when exposed to the decontaminating substance. It evaluates the significance of such reduced susceptibility in relation to the product efficacy, its impact on human and animal health, and the risk of ‘cross‐resistance’ to other biocides or therapeutic antimicrobials. Given the complexity and breadth of potential adverse effects – such as reduced susceptibility to the decontaminating substances under evaluation and to other biocides – and the absence of universally standardised guidelines for assessing biocide susceptibility, relevant information may need to be collected from diverse sources and interpreted in a case‐by‐case analysis. To enable a thorough evaluation, comprehensive information on reduced susceptibility to biocides and resistance to therapeutic antimicrobials should be provided, supported by established references and scientific literature.
These requirements also apply to substances or solutions with a so‐called ‘history of safe use’. An overview of the information requested can be found in Figure 1.
The standards and methodologies to be used in the experiments for chemical substances and biological agents as described in Sections 6.1 and 6.2, respectively, can be found in Section 6.3.
Framework for evaluating the potential emergence of reduced susceptibility to the decontaminating substance/formulated product, biocides and resistance to therapeutic antimicrobials. An overview of the required tests and decision‐making process is presented, incorporating the following elements: dark grey boxes indicate a process stop due to the identification of unacceptable concerns; grey boxes highlight areas where further tests or assessments are needed due to the presence of possible concerns or inconclusive prior knowledge; white boxes represent findings where acceptable or no concerns were identified, based on comprehensive evaluation, including the interpretation of specific data, supported by established references and the latest scientific knowledge provided by the applicant. The definitions for the terms we used have been included in the glossary.
Chemical substance(s)
6.1
Literature review for assessing the potential for emergence of reduced susceptibility to the chemical substance and/or other biocides and/or resistance to therapeutic antimicrobials
6.1.1
Information should be provided on the potential of the chemical substance and the formulated product to lead to stable or persistent development of, or selection for, reduced susceptibility to the substance/formulated product itself and/or to other biocides and/or resistance to therapeutic antimicrobials, including but not limited to, working concentrations and sub‐inhibitory concentrations of the substance on the target and non‐target species, including both Gram‐positive and Gram‐negative bacteria of human relevance, e.g. Escherichia coli, enterococci and Staphylococcus aureus. This should be based on up‐to‐date extensive literature review. Available data should be provided for decontamination applications, not limited to food (e.g. food processing environment or medical settings).
As presented in Figure 1, if unacceptable concerns are identified, the evaluation process is discontinued. For chemical substances and/or formulated products where prior knowledge is insufficient or inconclusive (e.g. insufficient number of representative strains tested), information should be provided as described in Sections 6.1.2, 6.1.4. In case acceptable or no concerns are identified, further assessments are not necessary at this stage. A plan for the post‐market evaluation should be provided (see Section 6.1.5).
Laboratory tests for assessing the emergence of reduced susceptibility to the chemical substance
6.1.2
Laboratory tests are required to evaluate the potential of the chemical substance(s) and formulated product(s) to select for acquired reduced susceptibility to the chemical substance and/or formulated product(s).
The initial screening should include laboratory testing, focusing on detecting emergence of reduced susceptibility to the chemical substance and/or formulated product in bacteria by creating experimental scenarios that closely mirror actual food surfaces targets, including potential sublethal dilutions of active substances, short exposure times, and the specific environmental conditions of the processing environment (e.g. temperature). The testing should include control and/or publicly available strains from culture collections of target pathogens (e.g. Campylobacter spp., Salmonella enterica, Listeria monocytogenes and Staphylococcus aureus) along with indicator (diverse Gram‐positive and Gram‐negative bacteria, such as E. coli and enterococci strains with different genetic and epidemiologic backgrounds) and other non‐target microorganisms with relevance to the processing environment. The minimum number of strains to be tested is 10 per species for each of the target pathogens and the indicator organisms and 10 across multiple species for other relevant non‐target microorganisms. The susceptibility tests for determining minimum inhibitory concentration (MIC) and minimal bactericidal concentration (MBC) should be performed. When possible, data supporting permanent changes in the bacterial susceptibility should be provided. In vitro evolution studies using serial passage experiment or time‐kill assays are also requested. When available, it is recommended to use an appropriate neutralisation method for the tests to investigate changes in the bacterial susceptibility to biocides (e.g. MBC, serial passage, time‐kill assay).
The initial screening should be validated in the processing plant or by using equivalent settings (such as pilot plants) under real conditions of decontaminant solution application. The in‐plant screening should evaluate the susceptibility profiles of microbial strains obtained. This evaluation would focus on any remaining strains, whether target or other relevant microorganisms (see Section 6.3), recovered from independent samples of food (e.g. carcass) after exposure to the formulated product. It is acknowledged that if the decontamination process is 100% effective, recovery of such strains may not be possible. Any recovered strain should be tested for their MIC and MBC against both the chemical substance(s) and the formulated product(s).
Analysis of ‘cross‐resistance’ and ‘co‐resistance’ between the chemical substance and other biocides and/or therapeutic antimicrobials
6.1.3
If acquired reduced susceptibility to the substance or formulated product is observed, further testing should assess whether these strains exhibit reduced susceptibility or resistance to other antimicrobials due to a common mechanism (‘cross‐resistance’). Additionally, it should evaluate whether the resistance mechanisms to the substance or formulated product are encoded on a genetic platform that also carries distinct resistance genes, conferring reduced susceptibility to other biocides and/or resistance to therapeutic antimicrobials (‘co‐resistance’).
For bacteria capable of acting as human pathogens, if identified in Section 6.1.2, the evaluation should specifically address ‘co‐resistance’ and ‘cross‐resistance’ with biocides as well as representatives of therapeutic antimicrobial classes.
If possible concerns are identified in the decreased susceptibility/resistance profiles assessed across various contexts, genetic analysis is required as explained in Section 6.1.4. If unacceptable concerns (e.g. ‘cross‐resistance’ to therapeutic antimicrobials) are identified the evaluation process is discontinued. In case acceptable or no concerns are identified, further assessments are not necessary at this stage. A plan for the post‐market evaluation should be provided (see Section 6.1.5).
Genetic analysis
6.1.4
The genetic analysis of microorganisms with decreased susceptibility to biocides and resistance to therapeutic antimicrobials is important for comprehensive understanding of the molecular mechanisms involved. This includes investigating the genetic basis of decreased susceptibility, such as point mutations in resistance‐associated genes, acquisition of resistance genes or changes in gene expression (please see outlined recommendations in Section 6.3).
In cases where genetic analysis reveals no molecular mechanisms of concern (such as intrinsic genes or transient gene overexpression), a post‐market evaluation and long‐term monitoring should be presented (as outlined in Section 6.1.5).
Post‐market evaluation and long‐term monitoring
6.1.5
To ensure the continuous safety of the chemical substance(s) or formulated product(s) a post‐market resistance surveillance plan must be provided, covering the aspects mentioned in the in‐plant screening as mentioned in Section 6.1.2. Additionally, data from monitoring conducted in countries or regions where the product is already on the market (e.g. other EU Member States or non‐EU countries) must be submitted.
If the decontamination solution is released into the environment without neutralisation, a post‐market monitoring plan is to be provided that could be used to assess the long‐term effects of using the chemical substance or formulated product on selection and dissemination of acquired reduced susceptibility to the chemical substance, other biocides and/or resistance to therapeutic antimicrobials in the environment. A statistically significant number of environmental samples should be collected in the wastewaters and both upstream and downstream of the point of discharge. The sampling strategy should take into account the seasonal changes and characteristics of the effluent. From the environmental samples taken, relevant indicator microorganisms should be isolated, identified, and used for monitoring of acquired reduced susceptibility to biocides and/or resistance to therapeutic antimicrobials, as described above.
Biological agent(s)
6.2
For a biological agent present in the decontamination solution, the applicant is referred to the EFSA guidance on the characterisation of microorganisms in support of the risk assessment of products used in the food chain (EFSA Scientific Committee, 2025a), more in particular to Section 3.2 (antimicrobial resistance) for the assessment of genes conferring resistance to therapeutic antimicrobials and to Section 3.3 (antimicrobial production). For bacteriophages, the search should be performed both in the genome of the bacteriophage itself (antimicrobial resistance) and in that of the production strain (antimicrobial resistance and antimicrobial production). The search should include any extrachromosomal element present in the strain under assessment.
It is also crucial to address the potential emergence of bacterial resistance to phages or phage cocktails and the co‐selection of resistance to therapeutic antimicrobials to ensure the long‐term efficacy and safety of this approach.
Literature review for assessing the potential for emergence of resistance to the biological agent and therapeutic antimicrobials or reduced susceptibility to chemical biocides
6.2.1
Information should be provided on the coevolution of bacteriophages and target bacteria, fitness costs of resistant bacteria, the risk of emergence and spread of resistant bacteria, and the molecular genetic basis of resistance. These studies should have been conducted with formulated products and the biological agent in vitro and/or in situ, including but not limited to, working conditions (e.g. phage concentrations, exposure times, and environmental factors). This should be based on up‐to‐date extensive literature review.
As presented in Figure 1, if unacceptable concerns are identified (e.g. permanent resistance to therapeutic antimicrobials induced by exposure to bacteriophages), the evaluation process is discontinued. For formulated products and/or biological agents where prior knowledge is insufficient or inconclusive (e.g. insufficient number of representative strains tested), information should be provided as described in Sections 6.2.2, 6.2.4. In case acceptable or no concerns are identified, further assessments is not necessary at this stage. A plan for the post‐market evaluation should be provided (see Section 6.2.5).
Laboratory tests for assessing the potential emergence of resistance to the biological agent
6.2.2
Laboratory tests are required to evaluate the potential of the biological agent and/or formulated product(s) to promote the emergence of resistance in the target strain(s) to the agent. Various conditions, including different phage concentrations, exposure times, and environmental factors should be tested.
The initial screening should include laboratory testing, simulating realistic processing environments, examining how the bacteriophage and its formulated products interact under diverse conditions such as varying concentrations, pH levels, temperatures, and exposure durations. Screening for emergence of phage resistant bacteria is to be performed by exposing bacterial susceptible cultures of the target strain(s) (including, control strains and/or publicly available strains from culture collections). Moreover, the stability and persistence of phage‐resistant strains in the absence of phage pressure is to be demonstrated. A representative subset of identified phage‐resistant bacterial isolates will need to be subjected to phenotypic characterisation, including the assessment of their growth kinetics and virulence potential.
The initial screening should be validated in the processing plant, if feasible, or by using equivalent settings (such as pilot plants) under real conditions of decontaminant solution application. This in‐plant screening is to be conducted by sampling target strains from the treated food and testing for their susceptibility to the applied phages or phage cocktails.
Analysis of ‘cross‐resistance’ and ‘co‐resistance’ between the biological agent and other biocides and/or therapeutic antimicrobials
6.2.3
Phenotypic characterisation of identified phage‐resistant target bacterial isolates (see Section 6.2.2) is to be performed. This includes assessing their potential for ‘cross‐resistance’ and ‘co‐resistance’ to therapeutic antimicrobials and to chemical biocides (to be used in the industrial settings where the agent is intended to be used) through MBC or MIC determination. In the presence of possible concerning findings based on these tests, genetic analysis is required as explained in Section 6.2.4. If unacceptable concerns (e.g. ‘cross‐resistance’ to therapeutic antimicrobials) are identified the evaluation process is discontinued. In case acceptable or no concerns are identified, further assessments are not necessary at this stage. A plan for the post‐market evaluation should be provided (see Section 6.2.5).
Genetic analysis
6.2.4
The mechanisms by which bacteria develop resistance to phages and, if applicable, to therapeutic antimicrobials and/or develop reduced susceptibility to biocides are to be examined using WGS or other appropriated methods. In addition, the potential for horizontal gene transfer and spread of resistance determinants is to be assessed when a resistance to therapeutic antimicrobials is identified (please see outlined recommendations in Section 6.3).
In cases where genetic analysis reveals no molecular mechanisms of concern (such as intrinsic genes or transient gene overexpression), a post‐market evaluation and long‐term monitoring should be presented (as outlined in Section 6.2.5).
Post‐market evaluation and long‐term monitoring
6.2.5
To ensure the continuous safety of the biological agent or formulated product(s) post‐market launch, a resistance surveillance plan should be provided aiming to regularly sample and test target bacterial strains isolated from treated independent samples of the intended food for their susceptibility to the applied phages or phage cocktails. Additionally, data from monitoring conducted in countries or regions where the product is already on the market (e.g. other EU Member States or non‐EU countries) must also be submitted.
If the decontamination solution is released into the environment without neutralisation, a structured monitoring plan is to be provided that could be used to assess the long‐term development of decreased susceptibility to chemical biocides and/or resistance to therapeutic antimicrobials and the biological agent/formulated product in the environment. A statistically significant number of environmental samples should be collected in the wastewaters and both upstream and downstream of the point of discharge. The sampling strategy should take into account seasonal changes and characteristics of the effluent. From the environmental samples taken, relevant indicator microorganisms should be isolated, identified and used for monitoring of acquired reduced susceptibility to biocides and/or resistance to therapeutic antimicrobials as described above.
Standards and methodologies
6.3
The standards and methodologies to be used in the experiments described in Sections 6.1 and 6.2 are as follows.
General principles
6.3.1
For in vitro susceptibility testing, reproducible and validated methods should be used with appropriate controls. When available, standardised methods, such as those from the International Organization for Standardization (ISO), European Committee on Antimicrobial Susceptibility Testing (EUCAST) or the Clinical and Laboratory Standards Institute (CLSI), should be applied to determine the MBC and MIC. However, as MICs and MBCs determined using these standardised methods may not fully capture resistance development under real‐world conditions of decontaminant use, it is recommended to also perform these tests under real‐world conditions or those identified in research as the most sensitive for detecting decreases in susceptibility (e.g. presence of organic load, short exposure times, temperature variability, food surface matrices). Each strain should be tested in triplicate to assess reproducibility.
For therapeutic antimicrobials susceptibility testing should be performed according to the EFSA Guidance on the characterisation of microorganisms in support of the risk assessment of products used in the food chain (EFSA Scientific Committee, 2025a).
When testing isolates during the in‐plant screening of the substance(s) or formulated product(s), the conditions of test or application shall be thoroughly documented, including presence of organic load, exposure time and environmental conditions.
For phages, only the target bacterial strains shall be tested, both during the initial screening and in‐plant screening, using plaque assay and/or planktonic killing assay.
Susceptibility testing interpretation
6.3.2
Susceptibility interpretation for therapeutic antimicrobials should follow the EFSA Guidance on the characterisation of microorganisms in support of the risk assessment of products used in the food chain (EFSA Scientific Committee, 2025a).
For biocides and decontaminants, MIC, MBC and any observed changes in susceptibility should be reported, using reproducible and validated methods; test conditions must be clearly documented. Where available, published ECOFFs or reference MICs should be used as benchmarks for interpretation. Where such benchmarks are not available, the interpretive criteria should be justified and it should be demonstrated that the methodology is appropriate for detecting changes in susceptibility under relevant conditions of use.
In vitro evolution studies
6.3.3
For in vitro evolution studies, the following applies:
- For serial passage experiments, bacterial cultures are to be exposed to sub‐lethal concentrations (including the highest possible sub‐lethal concentrations) of the substance(s)/agent(s) and formulated product(s) over multiple generations and be monitored for changes in resistance. This can help identify any evolutionary adaptations that confer increased resistance.
- For time‐kill assays, the bactericidal activity of the product is to be assessed over time, determining if bacterial populations can recover or grow in the presence of the biocide.
Characterisation of resistant strains
6.3.4
Where resistant or tolerant phenotypes are detected, additional studies should be carried out to assess their properties and potential impact.
Fitness cost assessment of phage‐resistant strains, growth rates of parent and resistant strains to phages should be compared and competition assays between parent and phage‐resistant strains performed to assess whether phage resistance imposes fitness costs on the target bacterial strain, as this can affect their virulence and survival in different environments.
For genetic and molecular analysis, WGS should be applied before and after exposure to identify mutations or acquired resistance genes. This is crucial for understanding the molecular basis of resistance and for the detection of resistance genes spread. Regarding the identification of acquired genes mediating resistance to therapeutic antimicrobials the applicant will adhere to the guidelines provided by EFSA (EFSA BIOHAZ Panel, 2023). Polymerase chain reaction (PCR) and quantitative PCR (qPCR) are to be used to track specific resistance genes within the bacterial population. qPCR can quantify the prevalence of resistance genes over time. Other advanced methodologies, such as Reverse Transcription Polymerase Chain Reaction (RT‐PCR) and transcriptomic analysis, may be applied as needed to complement these approaches and provide further insights into resistance mechanisms.
ENVIRONMENTAL RISK ASSESSMENT
7
Chemical substance(s)
7.1
The aim of environmental risk assessment (ERA) is to evaluate the potential for substances to affect the environment. In order to assess the safety of use of substances for the removal of microbial surface contamination of foods in the ERA, information is required about the conditions of application (see Section 3.3) as well as the release of the substances and their reaction products into the environment.
The ERA is based on the same principles as mentioned in the EFSA Guidance on the environmental risk assessment of feed additives (EFSA FEEDAP Panel, 2019), smoke flavours (EFSA FAF Panel, 2021) and the ECHA Guidance on the environmental risk assessment of biocides and industrial chemicals (ECHA, 2008a, 2017a). In the assessment, a predicted environmental concentration (PEC) is compared with a predicted no effect concentration (PNEC) value for each compartment of concern (water, sediment and soil). PNEC values are established from experimentally determined or predicted endpoints to which an appropriate assessment (safety) factor is applied. The value of the assessment factor (AF) is dependent on the amount of accurate and relevant data available, associated uncertainties and harmonisation requirements between different legislations (see Section 7.1.3 Environmental hazard assessment).
The workflow to be followed by the applicant in order to perform the ERA of the chemical substances used for the decontamination is presented below.
Data requirements
7.1.1
To assess the fate of the decontaminating substance(s), the minimum data requirements include data on physical–chemical properties, including water solubility, octanol/water partition coefficient (log K ow), vapour pressure and ionisation potential (see details in Section 3.1.1). Adsorption/desorption screening (according to OECD TG 106) is also needed, unless, based on the physico‐chemical properties of the substance, it can be expected to have a low potential for adsorption (e.g. when it has a low log K ow) or the substance and its relevant reaction products decompose rapidly. In addition, a Ready Biodegradability study (according to OECD TG 301) should be provided, unless high reactivity and/or rapid hydrolysis can be demonstrated. The information must cover the substance and all the relevant reaction products.
For the hazard assessment, the standard requirements are toxicity tests covering the three aquatic taxonomic groups: algae, invertebrates, and fish. The assessment should start with conducting acute toxicity tests according to OECD TG 201, 202 and 203, respectively. However, when a specific mode of action is expected (e.g. endocrine disruption), chronic toxicity tests should be conducted or used (when already available) to determine the hazard more accurately. Regarding the algal test (OECD TG 201), assays with green algae and with cyanobacteria are required for a proper assessment; if read‐across or another method clearly indicates that one taxonomic group is expected to be more sensitive than the other, the assay could be limited to the most sensitive taxon.
To assess the potential impact on the microbial function of the STPs, an activated sludge respiration inhibition test (according to OECD TG 209) is required, unless the substance is found to be readily biodegradable in a Ready Biodegradability study (according to OECD TG 301) and the applied test concentrations are in the range that is expected in the influent of a STP.
If it can be demonstrated that the substance will be completely transformed15 during the application and/or during treatment in a STP, the toxicity tests are not required and no environmental risk assessment is needed. This also applies to endogenous substance(s) (e.g. lactic acid) of which the concentration and/or distribution in the environment will not significantly be altered due to the decontamination treatment.
The generation of data using non‐testing approaches, such as (Q)SAR and read‐across, could also be considered to replace the tests mentioned above, provided the QSAR models are scientifically valid (using OECD principles, OECD, 2014) and adequate, and reliable documentation is provided. For an acceptable (Q)SAR prediction, the input must be correct, the substance fall within the applicability domain of the model, the prediction be reliable and the outcome fit for the regulatory purpose. The validity of models and predictions can be assessed by using the OECD (Q)SAR assessment framework (QAF) (OECD, 2023). Data on the possible environmental impact can be used from different sources, e.g. literature or other regulatory frameworks, provided that the original data are submitted to EFSA and the ownership of the data is guaranteed. Finally, all the available toxicological data on the substance should be considered in the assessment as they may, for instance, inform on the potential ecotoxicity.
Environmental exposure assessment
7.1.2
For the environmental exposure assessment, the emission scenario document developed for the biocidal use of disinfectants used in food and feed areas (JRC, 2011) can be followed to determine the release to the environment. In this document, it is assumed that untreated wastewater from slaughterhouses has a high organic load and is usually pretreated before release to the environment. Before the release into the sewer system, the wastewater is usually collected in tanks, where the pH is adjusted to a neutral range. It is assumed that wastewater of one slaughterhouse is released to one STP.
In accordance with this document, the release of substances used to remove surface contamination of foods to wastewater is considered by default 100%, but can be reduced if data are available justifying such a reduction. The disintegration of a substance during or after application and elimination during pretreatment of the wastewater before release to a STP are by default 0%, but can be increased if respective data are available (e.g. data on on‐site pretreatment of wastewater before release to the sewer system).
To calculate the PEC in the STP and the aquatic environment, the release to wastewater (expressed in kg/day) can be used as input for the model calculation described in chapter 2.3 of the Guidance on the Biocidal Products Regulation (ECHA, 2017a). If the physical–chemical properties and/or environmental fate studies indicate a potential of the substance or its transformation products to bind to STP sludge and/or sediment, the assessment should be extended to soil, covering the application of sewage sludge in agricultural soil and sediment.
Environmental hazard assessment
7.1.3
In the Guidance on the Biocidal Products Regulation (ECHA, 2017a), the REACH guidance document (ECHA, 2008a) and the EFSA Guidance for feed additives (EFSA FEEDAP Panel, 2019), a detailed description is provided on deriving a PNEC for the aquatic organisms and microorganisms in a STP. If test data are lacking, the equilibrium partitioning method can be used to derive a PNEC for soil and sediment organisms.
Environmental risk assessment
7.1.4
The risk assessment is based on a tiered approach. If the ratio PEC/PNEC is < 1, no further assessment is required, unless the substance has a log K ow ≥ 3. In that case, also the risk for secondary poisoning in the food chain has to be assessed, following the methodology outlined in the Guideline on environmental impact assessment for veterinary medicinal products in support of the VICH guidelines GL6 and GL38, Rev. 1 (EMA, 2016) and REACH Regulation (ECHA, 2008a, 2008b, 2016a, 2016b, 2023) and subsequent amendments.
The assessment of secondary poisoning through the aquatic/terrestrial food chain involves these key steps:
- Assessment of the potential for aquatic/terrestrial bioaccumulation and estimation of the exposure of top predators (calculation of concentration of substance in the food of the top predator (fish/earthworm), PEC_oral,predator_)
- Toxicity assessment (calculation of PNEC_oral_ from chronic oral and dietary toxicity studies)
- Risk characterisation (PEC_oral,predator_/PNEC_oral_)
If the PEC/PNEC is ≥ 1, a more refined PEC can be calculated based on additional data. If the PEC/PNEC ratio is still ≥ 1, a more refined PNEC, based on chronic toxicity data, can be derived (for further details, see the Guidance on the Biocidal Products Regulation, ECHA, 2017a) and Chapter 10 of the REACH guidance document (ECHA, 2008a).
Assessment of persistent, bioaccumulative and toxic and/or very persistent and very bioaccumulative substances
7.1.5
Special attention should be paid to substances that have the potential to be persistent, bioaccumulative and toxic (PBT) or very persistent and very bioaccumulative (vPvB). For them, there is more uncertainty in the estimation of risk and, as a consequence, a safe concentration in the environment cannot be established. Therefore, a separate hazard‐based PBT/vPvB assessment is required. To fulfil the PBT and vPvB assessment and to ensure a harmonised approach, the criteria as laid down in Annex XIII of the REACH Regulation (EC) No 1907/2006,16 together with the methodology in the current REACH Guidance on PBT assessment (ECHA, 2017b, 2023), should be considered.
The first step consists in screening whether the substance may fulfil these criteria. Screening information listed in appendix E of the Guidance on the safety assessment of feed additives for the environment (EFSA FEEDAP Panel, 2019) is recommended for comparing the screening information with screening thresholds (screening criteria) established for this purpose (for more details, see ECHA Guidance Chapter 11 on PBT/vPvB assessment, ECHA, 2017a) and ECHA Guidance on information requirements and chemical safety assessment Part C (ECHA, 2017b), Section C.4.1.
Substances considered to be potential PBT and/or vPvB on the basis of the screening assessment need to be further assessed with the PBT and vPvB criteria according to section 1 of Annex XIII of the REACH Regulation. Following the strategy outlined in the guidance documents, a definitive assessment of P/vP should be conducted first.
A definitive assessment of P/vP should normally be based on degradation half‐life data collected under adequate conditions for the relevant compartment(s) of exposure. If the substance is considered to fulfil the P and/or vP criterion, the PBT/vPvB assessment is continued by the evaluation of the B/vB criterion, including the assessment of newly generated additional information.
Definitive assessment of B/vB should normally be based on measured data on bioconcentration in aquatic species. If such data are not yet available, it is recommended to conduct a bioaccumulation study in fish according to OECD TG 305. If the substance is not identified as vPvB, but considered to fulfil the P (Persistent) and B (Bioccumulative) criteria, the PBT assessment is continued by an evaluation of the toxicity, based on the standard aquatic toxicity studies. The final assessment of T(Toxic) criterion should be based on the evaluation of the data for the classification of the substance for human health hazards and/or on no observed effect concentration/effect concentration of 10% (NOEC/EC_10_) values from long‐term toxicity tests with aquatic organisms.
It should also be considered whether the substances meet the criteria to be classified under the new hazard classes (persistent, mobile and toxic (PMT), very persistent and very mobile (vPvM) and endocrine disrupting (ED)) under the CLP Regulation No 1272/2008.17 There are transitional periods from the entry into force of this regulation.18 At the end of these transitional periods, all manufacturers, importers, downstream users and distributors must apply the new hazard classes. The updated guidance document to assess whether a substance fulfils these new criteria is available on the ECHA website.19
Biological agent(s)
7.2
As mentioned before, among biological agents, to date, only bacteriophages can be considered. The applicant should refer to section 5 (Environmental Risk Assessment) of the EFSA Guidance on the characterisation of microorganisms in support of the risk assessment of products used in the food chain (EFSA Scientific Committee, 2025a), specifically sections 5.1 and 5.3 that covers the ERA of non‐genetically modified and genetically modified active agents, respectively, as for the scope of this guidance document, bacteriophages are considered as active agents. The environment considered is the receiving environment(s) (e.g. environmental microbiome) that is exposed to the bacteriophage.
An ERA following the principles as described in section 5.1 of above mentioned guidance document (EFSA Scientific Committee, 2025a) is needed for non‐genetically modified bacteriophages, which are not common members of microbiome(s) in the receiving environment(s). Non‐genetically modified bacteriophages carrying acquired AMR genes, genes coding for toxins and/or virulence factors, are considered to be a risk. For genetically modified bacteriophages, the ERA is conducted on a case‐by‐case basis. It aims at identifying and evaluating the potential adverse effects resulting from the newly introduced trait(s) in the genetically modified bacteriophages on the receiving environment(s). More information can be found in sections 5.1 and 5.3 of above‐mentioned guidance document (EFSA Scientific Committee, 2025a).
ABBREVIATIONSAFCFood additives, flavourings, processing aids and materials in contact with foodAMRantimicrobial ResistanceB/vBbioaccumulative/very bioaccumulativeBIOHAZBiological HazardsBMDLbenchmark dose lower confidence limitCASChemical Abstracts ServiceCEPFood Contact Materials, Enzymes and Processing AidsCLSIClinical And Laboratory Standards InstituteECHAEuropean Chemicals AgencyECOFFepidemiological cut‐offEDendocrine disruptingED_10_ effective concentration of 10%EMAEuropean Medicines AgencyERAenvironment risk assessmentEUCASTEuropean Committee On Antimicrobial Susceptibility TestingFAsFood AdditivesFCMFood Contact MaterialsFDAFood and Drug AdministrationFEEDAPAdditives and Products or Substances used in Animal FeedFSAFood Standard AgencyFSMSfood safety management systemGLPGood Laboratory PracticeHACCPhazard analysis and critical control pointsHBGVhealth‐based guidance valuesISOInternational Organization for StandardizationIUPACInternational Union of Pure and Applied ChemistryLOAELlowest observed adverse effect levellog K ow octanol/water partition coefficientMBCminimal bactericidal concentrationMICminimal inhibitory concentrationMOEmargin of exposureMOImultiplicity of infectionNOAELno‐observed‐adverse‐effect‐levelNOECno observed effect concentrationOECDOrganisation for Economic Co‐operation and DevelopmentP/vPpersistent/very persistentPBTpersistent, bioaccumulative and toxicPCRpolymerase chain reactionPEpopulation equivalentsPECpredicted effect concentrationPFUplaque‐forming unit(s)PMTpersistent, mobile and toxicPNECpredicted no effect concentrationQAF(Q)SAR assessment frameworkqPCRquantitative polymerase chain reactionQPSqualified presumption of safetyQSARquantitative structural–activity relationshipREACHRegistration, Evaluation, Authorisation And Restriction Of ChemicalsRPreference pointRPCDsraw primary commodity derivativesRPCsraw primary commoditiesRT‐PCRreverse transcription polymerase chain reactionRTEready‐to‐eatRVPRegistration of Veterinary ProductsSCScientific CommitteeSOPstandard operation procedureSTPsewage treatment plantTDItolerable daily intakeTTCthreshold of toxicological concernVICHInternational Cooperation on Harmonization of Technical Requirements for Registration of Veterinary Medicinal ProductsvPvBvery persistent and very bioaccumulativevPvMvery persistent and very mobileWGSwhole genome sequencing
GLOSSARY Absorption (oral) The uptake of a chemical after oral administration from the gut into the cells lining the gut wall by transcellular processes and/or into the blood or lymph by either transcellular processes through the gut epithelium and/or by a paracellular pathway(s). Because of potential presystemic elimination, absorption does not necessarily lead to systemic availability, but absorption is necessary for a chemical to become systemically available. Acceptable concerns This term used in Section 6 refers to data that consistently indicate that any variations in susceptibility are minimal, reversible, or fall within the range of normal biological variability. In these cases, the decontaminating substance is not expected to contribute to a public health risk. Acquired AMR gene A resistance gene novel for the strain under assessment, acquired through horizontal transfer, enabling the bacterial strain to survive or multiply in the presence of concentrations of an antimicrobial agent higher than those that inhibit the growth of the majority of wild type strains of the same species without this AMR gene. AMR genes could be integrated in the bacterial chromosome or harboured on a separate genetic element. Acquired reduced susceptibility to biocides The situation when a bacterium develops tolerance to higher bacteriostatic or bactericidal concentrations than phenotypically related bacteria of the original or ‘wild type’ strain. Adverse effect Change in the morphology/physiology, growth, development, reproduction or lifespan of an organism, system or (sub)population that results in an impairment of functional capacity, an impairment of the capacity to compensate for additional stress or an increase in susceptibility to other influences (IPCS/WHO, 2009). Antimicrobial An active substance of synthetic or natural origin that destroys microorganisms, suppresses their growth or their ability to reproduce in animals or humans, excluding antivirals and antiparasitic agents. Antimicrobial activity The inhibitory or lethal effect of a decontaminating substance in a formulated product or an antibiotic. Antimicrobial resistance The ability of microorganisms of certain species to survive or even to grow in the presence of a given concentration of an antimicrobial that is usually sufficient to inhibit or kill microorganisms of the same species (ECDC, EMEA, EFSA, SCENIHR, 2009). Of primary concern is the emergence of resistance to therapeutic antimicrobials, defined below. Benchmark dose (BMD) The benchmark dose is a dose level, estimated from the fitted dose–response curve, associated with a specified change in response in a treated group relative to the control group (background response), the Benchmark Response (BMR) (EFSA Scientific Committee, 2022). Biocides A chemical substance or biological agent intended to destroy, deter, render harmless or exert a controlling effect on any harmful organism by chemical or biological means (CX 53‐2003).20
Co‐resistance The presence of resistance to more than one class of antimicrobial in the same bacterial strain, as might occur when different resistance genes are found on the same plasmid. Cross‐resistance A single resistance mechanism confers resistance to different antimicrobials. An example is the aminoglycoside‐modifying enzymes, which may confer resistance to several members of the aminoglycoside family. Cross‐resistance can occur across different classes of agents – a result of either overlapping drug targets, as is the case with macrolides and lincosamides, or a drug efflux pump with a broad range of activity (i.e. capable of exporting drugs of different classes of drugs). Decontaminating substance Substances applied to reduce pathogenic microorganisms from the surface of products or food of animal origin. The decontaminating substances may be either chemical substances (e.g. carboxylic acids, peroxy acids, or proteins), biological agents (e.g. bacteriophages), or a combination thereof. Decontamination solution The ‘decontamination solution’ refers to the final form of the substance as applied to the food surface, which could also contain stabilisers and/or surface active agents. It could consist of either a manually prepared solution or commercially available solution (i.e. formulated product), diluted or undiluted, or a combination of both. Food Safety Management system (FSMS) Prerequisite programmes that are preventive practices and conditions including all GHP, as well as other practices and procedures such as training and traceability, that establish the basic environmental and operating conditions that set the foundation for implementation of HACCP‐based procedures (EU Commission Notice, 2022/C355/01), supplemented with control measures at critical control point(s), as appropriate, that when taken as a whole, ensure that food is safe and suitable for its intended use. The FSMS is also the combination of control measures and assurance activities. The latter aims at providing evidence that control measures are working properly such as validation and verification, documentation and record keeping (EU Commission Notice, 2022/C355/01). Formulated product The ready‐to‐use product for which authorisation is sought. Good Hygiene Practices (GHP) Fundamental measures and conditions applied at any step within the food chain to provide safe and suitable food. GHP include also good manufacturing practice(s) (GMP, stressing correct work methodologies e.g. correct dosage of ingredients, appropriate processing temperature, checking that packages are clean and non‐damaged), good agriculture practice(s) (GAP, e.g. use of water of appropriate quality for irrigation, all in/all out system in animal rearing), good veterinarian practice(s) (GVP), good production practice(s) (GPP), good distribution practice(s) (GDP) and good trading practice(s) (GTP) (EU Commission Notice, 2022/C355/01). Gene of concerns Gene known to contribute to the production of toxins, toxic metabolites, therapeutic antimicrobials, acquired genes conferring resistance to therapeutic antimicrobials. For active agents (such as bacteriophages), virulence factors are also included in this definition. Genetically modified microorganisms Microorganisms in which the genetic material has been altered in a way that does not occur naturally by mating and/or natural recombination. Health‐base guidance value (HBGV) Umbrella term for values that are established as the result of the risk assessment of chemical substances and provides guidance on the safe consumption of substances, taking into account current safety data, uncertainties in these data, and the likely duration of consumption. Depending on their nature and applications, an HBGV for oral exposure may be termed acceptable daily intake (ADI) (food additives, pesticides), tolerable upper intake level (UL) (nutrients), tolerable daily intake (TDI) (contaminants, food contact materials) or acute reference dose (ARfD) (EFSA Scientific Committee, 2021b). Insufficient prior knowledge This category in Section 6 applies when existing data (from published studies, surveillance reports, or historical use) are too limited to allow a robust assessment. In such cases, the available literature or testing might include an insufficient number of strains or experiments, or the studies available may not be fully representative of the diversity in target or non‐target microorganisms. Inconclusive prior knowledge This category in Section 6 applies when the available data are contradictory or borderline. For example, some studies may suggest an effect while others do not, or the observed changes in susceptibility fall near the defined statistical or biological thresholds without clear consistency. Intrinsic AMR gene Gene inherent to strains of a bacterial species that limits the action of antimicrobial agents thereby allowing them to survive and multiply in presence of the antimicrobial agents. An AMR gene is considered ‘intrinsic’ if it is shared by the vast majority of wild type strains of the same species (or subspecies) and restricted to those located on the chromosome.21
Margin of exposure (MOE) The MOE is the ratio between the reference point (e.g. no‐observed‐adverse‐effect level (NOAEL) or benchmark dose lower confidence limit (BMDL), EFSA Scientific Committee, 2022) for the critical effect, and the estimated exposure (dose or concentration) to a substance for a given population (EFSA Scientific Committee 2025b). Microbiome Microbial community in a particular environment constituted by all taxonomic entities and their metabolic and genomic elements. Microorganism Any microbiological entity, cellular or non cellular, capable of multiplication or of transferring genetic material, including viruses, viroids, and animal and plant cells in culture. For the purpose of this guidance document, microorganisms cover bacteria, yeasts, filamentous fungi, microalgae and other protists and viruses including bacteriophages. Monitor The act of conducting a planned sequence of observations or measurements of control parameters to assess whether a control measure is under control (EU Commission Notice 2022/C355/01). No‐observed‐adverse‐effect‐level (NOAEL) Experimentally identified highest dose of a substance (expressed as mg/kg body weight) that causes no adverse alteration of morphology, functional capacity, growth, development or lifespan of the target organism distinguishable from those observed in control (untreated) organisms of the same species and strain under the same defined conditions of exposure. Possible concerns This term used in Section 6 refers to situations where preliminary data or borderline findings suggest that there might be an impact on susceptibility – but the evidence is not strong or consistent enough to classify the concern as ‘unacceptable’. Reaction products Substances formed in the decontamination solution as products of chemical transformation, degradation and other processes. Reference point (RP) The dose of a substance at which a low but measurable adverse effect is observed in a toxicity study. Therapeutic antimicrobials Antimicrobials of medical and veterinary importance for humans and animals as defined by the World Health Organization (WHO) and World Organization for Animal Health (WOAH), respectively (‘medically important antimicrobials’, WHO, 2024, 22 and future updates) and ‘veterinary critically important antimicrobial agents, veterinary highly important antimicrobial agents and veterinary important antimicrobial agents’ (WOAH, 202423 and future updates).24
Threshold of Toxicological Concern (TTC) The TTC approach is a screening and prioritisation tool for the risk assessment of chemicals when hazard data are incomplete and human exposure can be estimated (EFSA Scientific Committee, 2019). For substances with exposures below their corresponding TTC values, the probability that they would cause adverse health effects is low. If the estimated exposure to a substance is higher than the relevant TTC value, a non‐ TTC approach is required to reach a conclusion on potential adverse health effects. Tolerable daily intake The tolerable daily intake (TDI) is an estimate of the amount of a substance/body weight, which is present but not added deliberately to food or drinking water, and which can be consumed over a lifetime without presenting an appreciable risk to health. Unacceptable concerns This category in Section 6 applies when there are findings that indicate a level or type of reduced susceptibility or resistance, which poses a significant risk to public health (see the description in Figure 1 where dark grey boxes denote process stops due to unacceptable concerns). Uncertainty General term referring to all types of limitations in available knowledge that affect the range and probability of possible answers to an assessment question. Available knowledge refers here to the knowledge (evidence, data, etc.) available to assessors at the time the assessment is conducted and within the time and resources agreed for the assessment (EFSA, 2019b). Vapour pressure The vapour pressure of a substance is defined as the saturation pressure above a solid or a liquid substance at constant temperature (REACH Guidance). For the ERA, this property allows determination of the volatility of a substance from an aqueous medium or soil, in terms of the Henry's Law constant and partition coefficient air/soil, respectively. Virulence factor A cellular structure, molecule or regulatory system that enables pathogens to cause disease in a host by contributing to colonise or invade a niche, evade or inhibit its immune response, the acquisition of nutrients, or by directly causing host damage (e.g. through toxins).
REQUESTOR
European Food Safety Authority
QUESTION NUMBER
EFSA‐Q‐2024‐00020
PANEL MEMBERS
Riccardo Crebelli, Maria Joao Aleixo da Silva, Konrad Grob, Claude Lambré, Evgenia Lampi, Maria Rosaria Milana, Marja Pronk, Gilles Rivière, Mario Ščetar, Georgios Theodoridis, Els Van Hoeck and Nadia Waegeneers.
MAP DISCLAIMER
The designations employed and the presentation of material on any maps included in this scientific output do not imply the expression of any opinion whatsoever on the part of the European Food Safety Authority concerning the legal status of any country, territory, city or area or of its authorities, or concerning the delimitation of its frontiers or boundaries.
Supporting information
ANNEX A: Report on the public consultation on the draft guidance document on the submission of data for the evaluation of the safety and efficacy of substances for the removal of microbial surface contamination of foods of animal origin intended for human consumption
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Cramer, G. M. , Ford, R. A. , & Hall, R. L. (1978). Estimation of toxic hazard – A decision tree approach. Food and Cosmetics Toxicology, 16(3), 255–276. 10.1016/s 0015-6264(76)80522-6 357272 · doi ↗ · pubmed ↗
- 2Dhowlaghar, N. , & Denes, T. G. (2023). Control of residual phage in the evaluation of phage‐based food safety applications. Critical Reviews in Food Science and Nutrition, 64, 1–7. 10.1080/10408398.2023.2258210 37707444 · doi ↗ · pubmed ↗
- 3ECDC, EMEA, EFSA, SCENIHR . (2009). Scientific Opinion of the European Centre for Disease Prevention and Control; Scientific Opinion of the Panel on Biological Hazards; Opinion of the Committee for Medicinal Products for Veterinary Use; Scientific Opinion of the Scientific Committee on Emerging and Newly Identified Health Risks on antimicrobial resistance (AMR) focused on zoonotic infections. EFSA Journal, 7(11), 1372. 10.2903/j.efsa.2009.1372 · doi ↗
- 4ECHA (European Chemicals Agency) . (2008 a). Guidance on information requirements and chemical safety assessment Chapter R.10: Characterisation of dose [concentration]‐response for environment . https://echa.europa.eu/documents/10162/17224/information_requirements_r 10_en.pdf/bb 902be 7‐a 503‐4ab 7‐9036‐d 866b 8ddce 69
- 5ECHA (European Chemicals Agency) . (2008 b). Guidance for the implementation of REACH, Chapter R.6: QSA Rs and grouping of chemicals . https://echa.europa.eu/documents/10162/13632/information_requirements_r 6_en.pdf/77f 49f 81‐b 76d‐40ab‐8513‐4f 3a 533b 6ac 9
- 6ECHA (European Chemicals Agency) . (2016 a). Guidance on Information Requirements and Chemical Safety Assessment Chapter R.16: Environmental exposure assessment. Version 3.0 . https://echa.europa.eu/documents/10162/17224/information_requirements_r 16_en.pdf/b 9f 0f 406‐ff 5f‐4315‐908e‐e 5f 83115 d 6af
- 7ECHA (European Chemicals Agency) . (2016 b). Practical guide. How to use and report (Q)SA Rs. Version 3.1, July 2016. https://echa.europa.eu/documents/10162/13655/pg_report_qsars_en.pdf/407dff 11‐aa 4a‐4eef‐a 1ce‐9300 f 8460099
- 8ECHA (European Chemicals Agency) . (2017 a). Guidance on biocidal products regulation: volume IV environment ‐assessment and evaluation (Parts B+C) . https://echa.europa.eu/documents/10162/23036412/bpr_guidance_ra_vol_iv_part_b‐c_en.pdf/e 2622 aea‐0b 93‐493f‐85a 3‐f 9cb 42be 16ae