Construction of remote dual stereocenters by electrochemical cobalt-catalyzed enantioselective desymmetrization
Yanjun Li, Siyu Liu, Binbin Yuan, Nico Graw, Lutz Ackermann

TL;DR
Scientists developed a new electrochemical method to create two distinct chiral centers far apart in a single step using cobalt catalysis.
Contribution
A novel electrochemical cobalt-catalyzed method for enantioselective desymmetrization enabling remote dual stereocenter formation.
Findings
An electrochemical cobalt-catalyzed asymmetric hydroacylation of enynes enables enantio- and diastereoselective construction of remote dual stereocenters.
The method produces four classes of chiral products with distinct stereogenic elements.
The approach demonstrates broad substrate generality and stereochemical control.
Abstract
The enantio- and diastereoselective construction of two stereogenic centers represents a highly attractive objective in synthetic chemistry. Extensive asymmetric catalytic methods have been developed for the formation of vicinal stereocenters. In contrast, the simultaneous construction of two constitutionally distinct stereogenic centers at remote positions in a single asymmetric catalytic step remains very scarce, owing to the lack of reliable models for distant stereochemical induction for both chiral entities. Herein, we report on an electrochemical cobalt-catalyzed asymmetric hydroacylation of enynes by a desymmetrization strategy that enables the enantio- and diastereo-selective construction of remote dual stereocenters. This unified catalytic platform exhibits broad substrate generality and affords four distinct classes of chiral products, each incorporating two chiral elements:…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
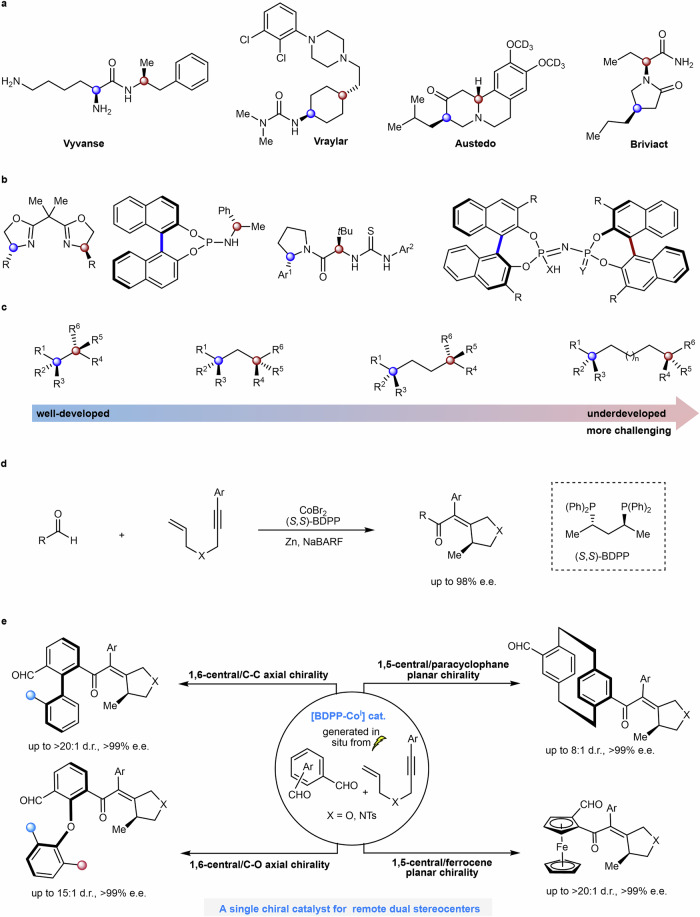 Figure 1
Figure 1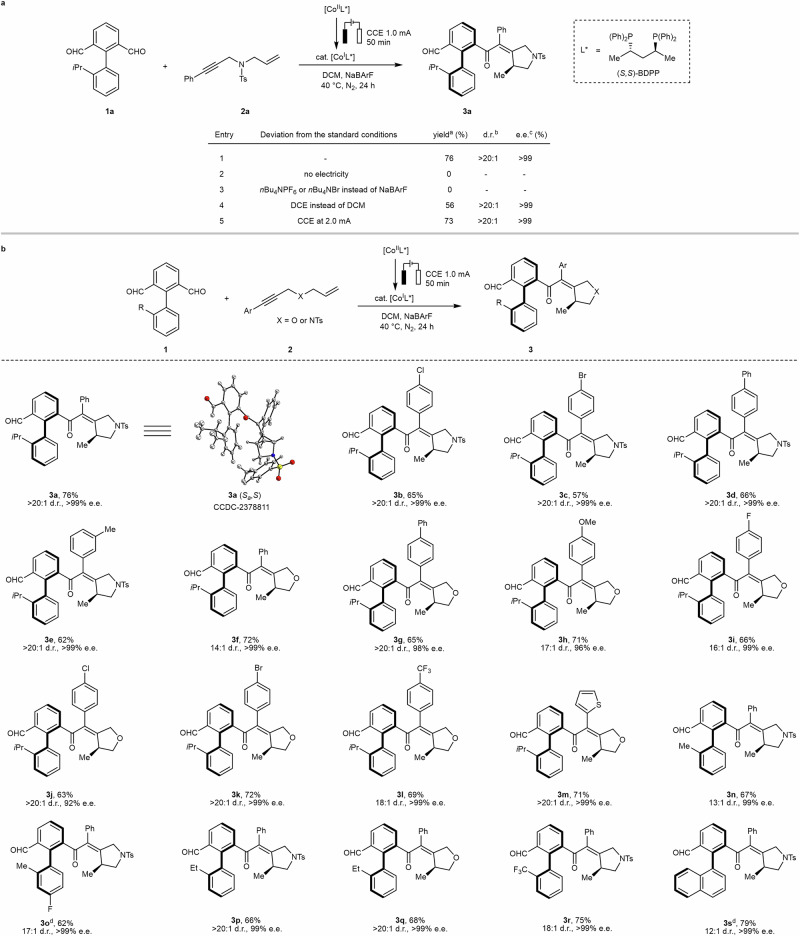 Figure 2
Figure 2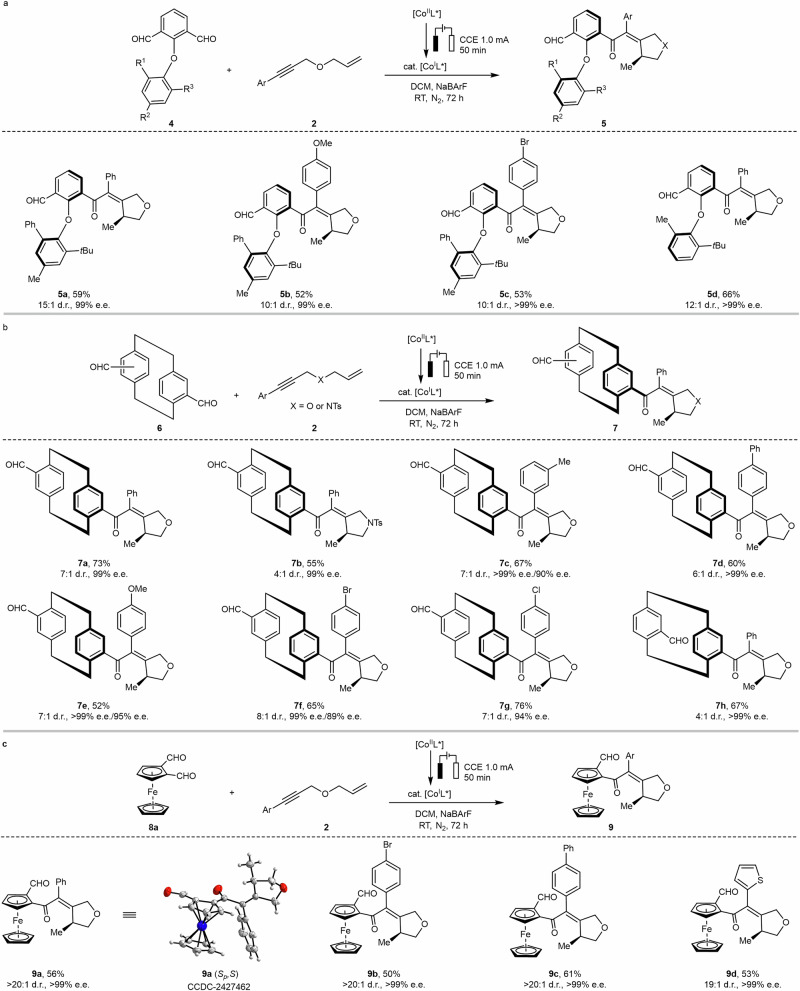 Figure 3
Figure 3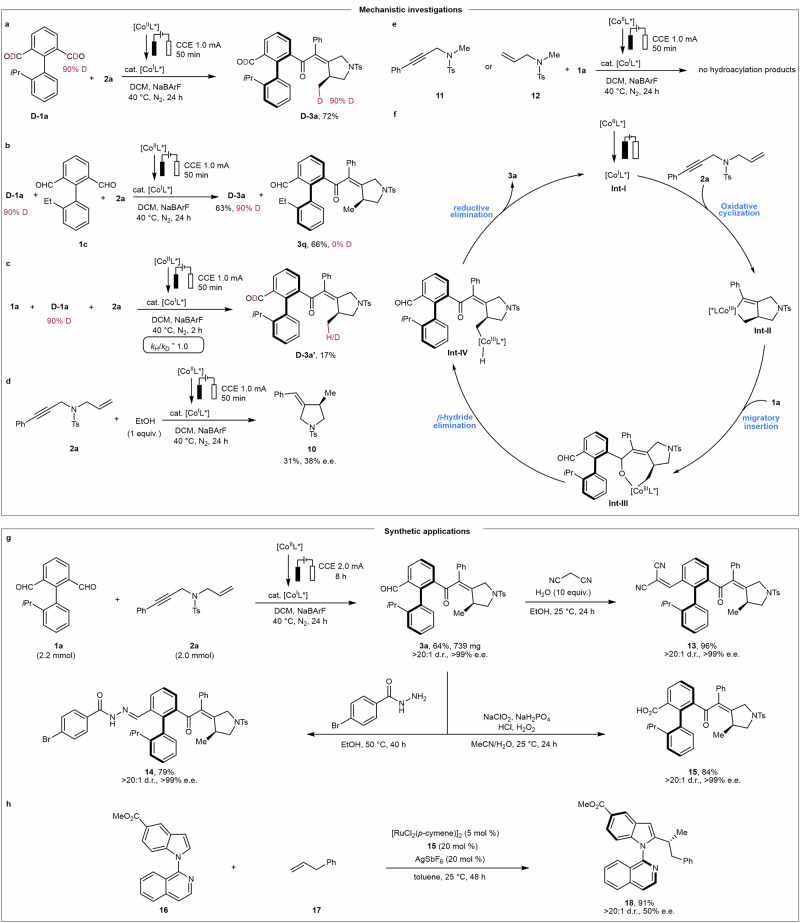 Figure 4
Figure 4- —https://doi.org/10.13039/501100001659Deutsche Forschungsgemeinschaft (German Research Foundation)
- —https://doi.org/10.13039/100010663EC | EU Framework Programme for Research and Innovation H2020 | H2020 Priority Excellent Science | H2020 European Research Council (H2020 Excellent Science - European
- —https://doi.org/10.13039/501100004543China Scholarship Council (CSC)
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsRadical Photochemical Reactions · Axial and Atropisomeric Chirality Synthesis · Catalytic C–H Functionalization Methods
Introduction
Asymmetric catalysis has achieved significant progress over the past decades. Numerous synthetic approaches have been developed for the enantio- and diastereoselective construction of molecules containing 1,2-diastereogenic centers^1–3^. Remote dual stereocenters are prevalent in chiral ligands/catalysts^4–8^ and chiral drugs^9^, including four of the top 100 drugs by retail sales in 2023 (Fig. 1a, b). However, catalytic construction of two nonadjacent and constitutionally distinct stereocenters has long remained a formidable challenge, particularly for 1,n-nonadjacent (n ≥ 5) stereocenters^10^. Furthermore, the larger the distance between two stereocenters, the more difficult it becomes to achieve asymmetric induction (Fig. 1c)^11^. The merger of synergistic catalysis that employs two distinct chiral catalysts to control double chiral induction at two 1,n-nonadjacent (n ≥ 5) stereocenters has only recently been demonstrated^10–13^. However, synergistic catalytic systems frequently face challenges such as incompatibility between multiple catalysts and substrates, as well as ensuring the correct sequence of each process, which greatly limits both the range of possible reactions and the diversity of products. The use of a single catalyst to control chiral induction at remote dual stereocenters bearing different chiral elements has thus far remained elusive^14^.Fig. 1. Motivation and strategy for the construction of remote dual stereocenters.a Examples of drugs bearing remote dual stereocenters (four of the top 100 drugs by retail sales in 2023). b Examples of ligands/catalysts bearing remote dual stereocenters. c The difficulty of one-step asymmetric construction of two stereocenters. d Lautens’s work: cobalt-catalyzed enantioselective intermolecular hydroacylation of 1,6-enynes. e This work: construction of remote dual stereocenters by electrochemical cobalt-catalyzed enantioselective desymmetrization. d.r. diastereoselectivity. e.e. enantiomeric excess. NaBArF sodium tetrakis[3,5-bis(trifluoromethyl)phenyl]borate.
Achieving substrate generality remains a formidable challenge in asymmetric catalysis^15–17^, particularly in the synthesis of products containing different chiral elements. Most asymmetric catalytic systems are effective only for a limited range of substrates and are typically confined to establishing a single type of chirality. Usually, accessing different chiral elements by asymmetric catalysis requires distinct synthetic strategies and substantial effort to optimize chiral catalysts and reaction conditions. Although catalytic strategies for the synthesis of compounds with two types of chiral elements^18^, such as central-axial^19–23^, central-planar^24^, central-helical^25^, axial-planar^26,27^, and axial-helical^21^, have been devised individually, it remains difficult to access a broader variety of chiral elements in a unified catalytic system because of their distinct topologies.
Transition-metal-catalyzed enantioselective hydroacylation of alkenes has emerged as a powerful strategy for the synthesis of chiral ketones through the asymmetric addition of aldehyde C–H bonds across alkenes^28–34^. Pioneering studies on cobalt-catalyzed enantioselective intramolecular hydroacylation of alkenes were achieved by Yoshikai^30,31^. Subsequently, Lautens disclosed a cobalt-catalyzed intermolecular hydroacylation of 1,6-enynes with aldehydes (Fig. 1d)^34^. Enantioselective desymmetrization transforms prochiral or meso substrates into chiral products by generating one or more stereogenic centers, providing efficient access to structurally complex molecules from readily available starting materials. This strategy has been widely applied in various transformations, including aldol reactions, epoxidations, reductions, cross-coupling reactions, and ring-closing metathesis^35–39^.
More recently, asymmetric electrochemical transformations have attracted considerable attention as a versatile platform for the construction of chiral molecules^40,41^. Within our continuous interest in electrochemical enantioselective C–H functionalization^22,42–44^, we have now identified the electrochemical cobalt-catalyzed asymmetric hydroacylation of enynes for the construction of remote dual stereocenters by a desymmetrization strategy, on which we report herein (Fig. 1e). This unified catalytic platform exhibits substrate generality and affords four distinct classes of chiral products, each containing two chiral elements. These include 1,6-central/C–C axial chirality, 1,6-central/C–O axial chirality, 1,5-central/[2.2]paracyclophane planar chirality, and 1,5-central/ferrocene planar chirality.
Results
Our investigation was initiated with the reaction of biaryl dialdehyde 1a with 1,6-enyne 2a. After intensive optimization studies (see details in Supplementary Table 1), the optimal conditions were identified as follows: a catalytic amount of [(S,S)-BDPP]CoBr_2_ was electrochemically reduced with a constant current of 1.0 mA in an undivided cell at 40 °C for 50 min; then substrates 1a and 2a were added, and the reaction mixture was stirred at 40 °C. The standard conditions afforded the desired product 3a in 76% yield with > 20:1 d.r. and > 99% e.e. (Fig. 2a, entry 1). The structure of product 3a was unambiguously confirmed by X-ray diffraction analysis. The atropostability of the C–C axis in product 3a was calculated via density functional theory, showing a rotational barrier of 32.7 kcal mol^−1^, indicative of a half-life t1/2 of 112.3 years (see Supplementary Information section 7 for details). Control experiments revealed that electricity was essential for the reaction (Fig. 2a, entry 2). When nBu_4_NPF_6_ or nBu_4_NBr were used as the electrolytes, no desired product was generated (Fig. 2a, entry 3). The choice of solvent was key to success, as evidenced by the low yield (0–56%) obtained when using other solvents (Fig. 2a, entry 4; Supplementary Table 1). At a constant current of 2.0 mA, a slightly decreased yield was noted (Fig. 2a, entry 5). Other chiral phosphine ligands were tested but failed to provide the desired product (see details in Supplementary Table 1).Fig. 2. Reaction development and establishing 1,6-central/C−C axial chirality.a Reaction development. Standard reaction conditions: [(S,S)-BDPP]CoBr_2_ (0.010 mmol), NaBArF (0.050 mmol), DCM (2.0 mL), undivided cell, constant current at 1.0 mA, zinc plate anode and nickel foam cathode, 40 °C, 50 min; then 1a (0.11 mmol), 2a (0.10 mmol), 40 °C, 24 h. ^a^Isolated yield. ^b^d.r. is determined by ^1^H-NMR. ^c^e.e. is determined by chiral HPLC analysis. ^d^At room temperature (RT) for 48 h. b The construction of 1,6-central/C−C axial chirality. DCM dichloromethane, DCE 1,2-dichloroethane, CCE constant current electrolysis.
With the optimized conditions in hand, the substrate robustness of the electrocatalysis was investigated (Fig. 2b). First, various substituents on the phenyl ring of 1,6-enynes 2 were evaluated. As a result, both electron-donating and electron-withdrawing groups were found to be well-tolerated, delivering the corresponding products with 1,6-central/C−C axial chirality (3a − 3 l) in good yields (57 − 76%) with excellent enantioselectivities (92% to > 99% e.e.) and good to excellent diastereoselectivities (14:1 to > 20:1 d.r.). Second, a substrate bearing a thiophene motif was successfully transformed into the corresponding product 3 m. Third, we probed the scope of different substituted biphenyl bialdehydes. The replacement of the isopropyl group at the 2-position of the benzene ring with methyl, ethyl, or trifluoromethyl substituent was feasible, giving the desired products 3n−3r in good yields with excellent enantioselectivities (up to > 99% e.e.) and good to excellent diastereoselectivities (13:1 to > 20:1 d.r.). The reaction of naphthyl-substituted dialdehyde with 2a efficiently yielded product 3 s with high enantio- and diastereoselectivities.
Encouraged by the results on C–C atropisomers, we were keen to expand this electrocatalytic system to the synthesis of more challenging atropisomeric diaryl ethers. Atropisomeric diaryl ethers with restricted C−O bond rotation are found in biological molecules and serve as promising frameworks for designing chiral ligands^45^. However, limited reported methods for the synthesis of chiral diaryl ethers have significantly hindered progress in the field when compared to well-studied axially chiral biaryls^45–49^. In this context, we envisioned that if a C–O axis-containing prochiral substrate is subjected to our strategy, the [(S,S)-BDPP]-cobalt catalyst has the propensity to modulate the C–O axis steric environment and spatial distribution enabling an efficient chirality control. This can be attributed to the structural characteristics of (S,S)-BDPP, which features a flexible chiral secondary carbon backbone with 2,4-bis(diphenylphosphino) substituents, allowing the catalyst to adopt a suitable P–Co–P coordination angle. Gratifyingly, under the standard reaction conditions, the diaryl ethers substrate with 1,6-enynes gave products 5a−5 d in 52 − 66% yield with excellent enantioselectivities (up to > 99% e.e.) and good to excellent diastereoselectivities (10:1 − 15:1 d.r.) (Fig. 3a).Fig. 3. Construction of 1,6-central/C–O axial chirality and 1,5-central/planar chirality.a The construction of 1,6-central/C–O axial chirality. b The construction of 1,5-central/[2.2]paracyclophane planar chirality. c The construction of 1,5-central/ferrocene planar chirality.
The above results inspired us to explore the wider substrate generality. Planar chiral [2.2]paracyclophane derivatives are fascinating chiral molecules with significant applications in asymmetric catalysis and materials science^50,51^. However, current synthetic methods to obtain enantioenriched [2.2]paracyclophanes remain largely reliant on chiral chromatography or chemical resolution, with only a few examples of asymmetric catalysis reported^51–54^. Asymmetric transformations of [2.2]paracyclophane derivatives are particularly challenging due to their intrinsic structural distortions, rigidity, and pronounced steric hindrance. As such, these compounds shine as challenging molecules, and therefore excellent target motifs to assess the robustness and versatility of our framework. Notably, the reaction of paracyclophane dicarbaldehydes 6 with 1,6-enynes 2 afforded the anticipated products (7a–7 h) featuring 1,5-central/planar chirality in 44–60% yield, with excellent enantioselectivities (up to > 99% e.e.) and moderate to good diastereoselectivities (4:1–8:1 d.r.) (Fig. 3b). It is noteworthy that some of the diastereoisomeric products (7c, 7e, 7f) can be easily separated by silica gel chromatography, and the minor diastereomers were obtained with good enantiomeric excess (89%–95% e.e.). The application of our approach was next extended to another typical planar chiral structure−chiral ferrocene. The ferrocene dicarbaldehydes with 1,6-enynes gave desired products 9a–9d with 1,5-central/planar chirality in 50–61% yield with excellent enantioselectivities ( > 99% e.e.) and diastereoselectivities (up to > 20:1 d.r.) (Fig. 3c). The structure of product 9a was confirmed by single-crystal X-ray diffraction. The introduction of remote dual stereocenters into [2.2]paracyclophane or ferrocene could further extend their potential applications, for instance in the design of chiral ligands.
In order to shed light into the catalyst’s mode of action, we carried out several mechanistic studies (Fig. 4). When deuterated dialdehyde D-1a (90% D) was subjected to the standard reaction conditions (Fig. 4a), the product D-3a was obtained with 90% deuterium incorporation. Deuterium crossover experiments with substrates D-1a and 1c revealed that deuterium was solely incorporated into D-3a (Fig. 4b). The absence of deuterium crossover rules out a ligand-to-ligand hydrogen transfer pathway. A KIE of kH/kD ≈ 1.0 was suggestive of the C−H cleavage not being involved in the turnover-limiting step (Fig. 4c). The results of deuterium incorporation and deuterium crossover experiments were consistent with Lautens’s work^34^. The reaction of enyne 2a in the presence of ethanol gave product 10, indicating that the reaction proceeds via a cobaltacycle species (Fig. 4d)^55^. Substrates 11 and 12, without either the alkyne or alkene moiety of the enyne, gave no hydroacylation products under the reaction conditions, being indicative of the oxidative cyclization of the enyne (Fig. 4e).Fig. 4. Mechanistic investigations and synthetic applications.a Deuterium labeling experiments. b Deuterium crossover experiments. c KIE experiments. d Control experiments for the reaction of enyne with ethanol. e Control experiments with alkyne or alkene. f Proposed mechanism. g Gram-scale synthesis and follow-up transformations of product 3a. h Chiral carboxylic acid 15 was evaluated as a chiral ligand in the ruthenium-catalyzed asymmetric C−H activation reaction.
Based on these experimental findings, a proposed catalytic cycle is depicted in Fig. 4f. The reaction initiates with the formation of the low-valent chiral cobalt(I) active catalyst Int-I via cathodic reduction of [(S,S)-BDPP]CoBr_2_^56^. Cyclic voltammetry assigned the cobalt(II)/cobalt(I) redox couple a potential of –0.56 V (see details in Supplementary Fig. 5), indicating that the low-valent chiral cobalt species is readily generated under electroreductive conditions, thereby supporting the proposed catalyst activation pathway. The latter upon coordination of the enyne 2a undergoes enantioselective oxidative cyclization to generate the cobaltacycle Int-II with the construction of the first stereogenic center. Then, migratory insertion of the dialdehyde 1a takes place, which enables the formation of the cobaltacycle Int-Ⅲ leading to the formation of the second stereogenic center. Thereafter, β-hydride elimination followed by reductive elimination delivers the desired product 3a as well as the regeneration of the catalyst. An alternative pathway contemplating oxidative addition is considered and depicted in Supplementary Fig. 6.
Thereafter, diverse synthetic transformations were performed to demonstrate the synthetic practicality of our methodology (Fig. 4g). Notably, a gram-scale electroreduction provided the product 3a in good yield with excellent enantioselectivity ( > 99% e.e.) and diastereoselectivity ( > 20:1 d.r.). The reaction of 3a with cyanoacetonitrile gave compound 13 in 96% yield. Condensation of the formyl group with benzohydrazide afforded the corresponding imine 14 in 79% yield. Oxidation of 3a afforded chiral carboxylic acid 15 in 84% yield. To our delight, all these transformations proceeded with the retention of both enantioselectivity as well as diastereoselectivity. Furthermore, chiral carboxylic acid 15 was further examined as a chiral ligand in the ruthenium-catalyzed asymmetric C−H activation, furnishing the product 18 with both central and axial chirality in moderate enantioselectivity and excellent diastereoselectivity (Fig. 4h).
Discussion
A uniquely robust and versatile strategy for remote dual stereocenters featuring diverse elements was viable through electroreductive cobalt-catalyzed enantioselective desymmetrization. Through the use of a single catalytic system and without any further optimization, our strategy enables the generation of various stereogenic elements, including central chirality, C–C/C–O axial chirality, [2.2]paracyclophane planar chirality, and ferrocene planar chirality. The mechanism demonstrates that the catalyst directly governs two stereoselectivities in different elementary steps in the catalytic cycle, thus enabling precise control over 1,5/1,6-double chiral inductions.
Methods
General procedure for electrochemical cobalt-catalyzed enantioselective desymmetrization
The electrocatalysis was carried out in an undivided cell, with a zinc electrode (10 mm × 25 mm × 0.25 mm) and a nickel foam electrode (10 mm × 25 mm × 1 mm). In the glovebox, [(S,S)-BDPP]CoBr_2_ (6.6 mg, 0.010 mmol), NaBArF (44.3 mg, 0.050 mmol), and dry DCM (2 mL) were placed in a 10 mL cell. Electrocatalysis was performed at 40 °C with a constant current of 1.0 mA maintained for 50 min. Then, the nickel foam cathode and the zinc anode were taken out, bialdehyde (0.11 mmol) and 1,6-enyne (0.10 mmol) were added, and the reaction mixture was stirred at 40 °C or room temperature under a nitrogen atmosphere for 24–72 h. The resulting mixture was purified by column chromatography on silica gel to afford the desired product.
Supplementary information
Supplementary Information Transparent Peer Review file
Source data
Source Data
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Carreira, E. M. & Yamamoto, H. (eds.) Comprehensive Chirality. (Academic, 2012).
- 2Akiyama, T. & Ojima I. Catalytic Asymmetric Synthesis. (Wiley, 2022).
- 3Zhou, Q.-L. et al. Privileged Chiral Ligands And Catalysts. (Wiley, 2011).
- 4Lin, C., Xu, X., Chong, Q. & Meng, F. Simultaneous construction of axial and central stereogenicity by cobalt-catalyzed stereoconvergent reductive coupling of heterobiaryl triflates and aldehydes. Chem. Eur. J. 31, e 202500248 (2025).10.1002/chem.20250024840008493 · doi ↗ · pubmed ↗