Terrible APS — a newly defined variant of severe APS
Stanley Niznik, Tania Zaher, Soad Haj Yehia, Ronen Shavit, Shiri Weinstein, Yulia Lifshitz, Nancy Agmon-Levin

TL;DR
This study introduces 'Terrible APS' (TrAPS), a severe and refractory variant of antiphospholipid syndrome, and proposes a scoring system to identify it.
Contribution
The paper defines a new APS subcategory, TrAPS, and introduces the TrAPScore for its diagnosis.
Findings
27 out of 209 thrombotic APS patients met the TrAPS criteria, showing higher rates of severe complications.
TrAPS was associated with increased mortality (18.5% vs. 5.1%) and anticoagulant resistance.
The TrAPScore, based on four predictors, effectively identifies TrAPS with high predictive values.
Abstract
Antiphospholipid syndrome (APS) presents with various clinical features and some patients exhibit progressive, refractory disease that does not meet the catastrophic APS (cAPS) criteria. This study describes a new subcategory of APS patients, “Terrible APS” (TrAPS), characterized by recurrent thrombosis despite optimal anticoagulation, often requiring immunomodulation or surgery. We analyzed 306 primary APS patients, excluding those with obstetric, non-criteria APS, or cAPS. TrAPS was defined as >2 breakthrough thrombotic events despite anticoagulation (without provocation or cardiovascular risk) or the need for >1 immunomodulatory or surgical intervention. Among 209 patients with thrombotic primary APS, 27 (12.7%) met the TrAPS criteria. These patients had higher rates of venous thrombosis, microvascular involvement, heart valve disease, thrombocytopenia, and triple-positive…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
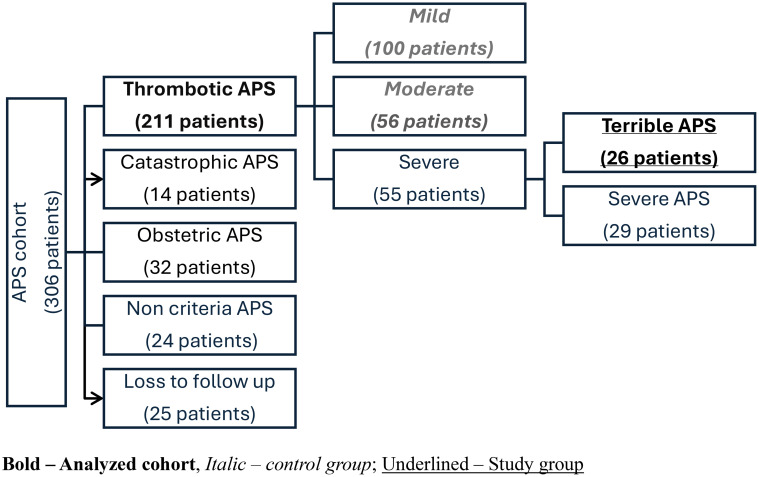 Figure 1
Figure 1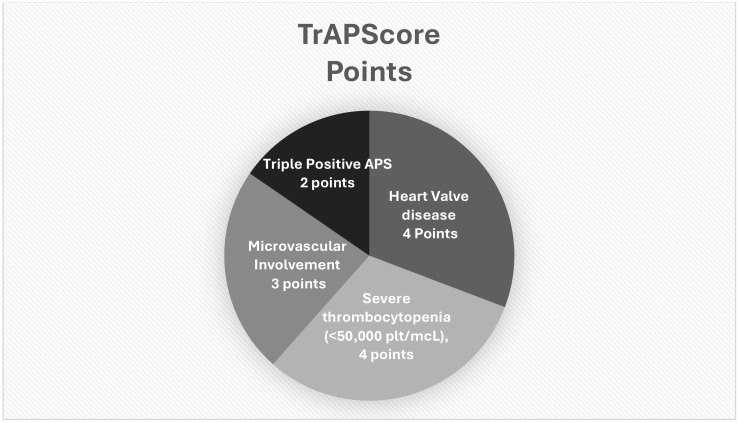 Figure 2
Figure 2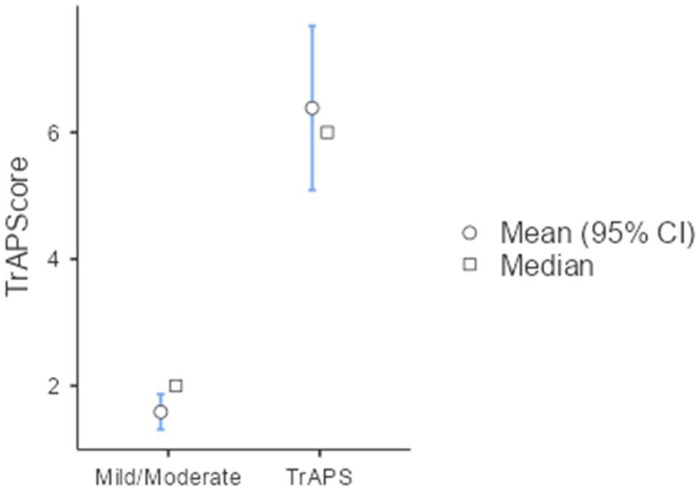 Figure 3
Figure 3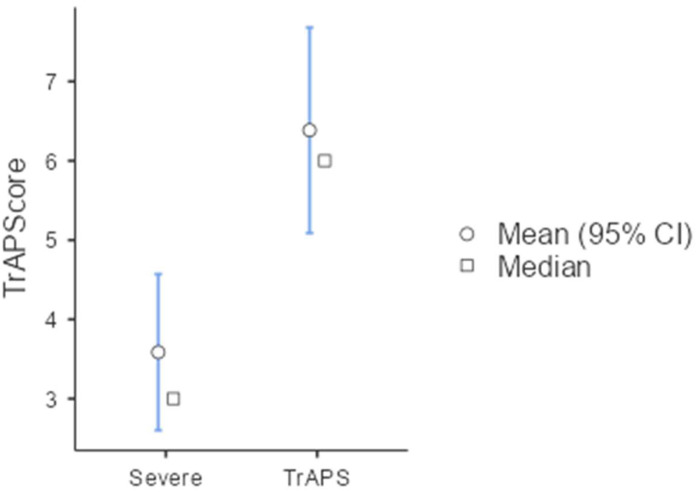 Figure 4
Figure 4| Parameter | Terrible APS (n = 26) | APS control group (n = 156) | P value |
|---|---|---|---|
| Gender (Female) | 20 (76.9%) | 110 (70.8%) | 0.708 |
| Age at presentation | 34 (CI95% 32–40.8) | 38 (CI95% 30.1–45.9) | 0.199 |
| Follow up period (years) | 16 (CI95% 10.3–18.45) | 11.1 (CI95% 6.6–15.6) | 0.012 |
| Secondary APS diagnosis during follow-up | 1 (3.8%) | 16 (10.2%) | 0.711 |
| Hypertension | 6 (23.1%) | 41 (26.2%) | 0.658 |
| Smoking | 5 (19.2%) | 30 (19.2%) | 0.588 |
| Dyslipidemia | 6 (23.1%) | 35 (22.4%) | 0.981 |
| Diabetes Mellitus | 2 (7.7%) | 12 (7.7%) | 0.959 |
| Obesity | 8 (30.7%) | 29 (18.8%) | 0.186 |
| Venous thrombosis at presentation* | 15 (57.7%) | 45 (28.8%) | 0.006 |
| Arterial Thrombosis at presentation | 16 (61.5%) | 78 (50%) | 0.21 |
| Obstetric morbidity at presentation (female only) | 4 (14.8%) | 50 (32%) | 0.07 |
| Parameter | Terrible APS (n = 26) | APS control group (n = 156) | P value |
|---|---|---|---|
| Anti B2 Glycoprotein IGG positivity | 24 (92.3%) | 96 (69.5%) | 0.005 |
| Average B2 Glycoprotein IgG Titer (u/ml) | 131 (± 56.3) | 68.6 (± 67.3) | <0.001 |
| Anti B2 Glycoprotein IgM positivity | 12 (46.1%) | 75 (48%) | 0.727 |
| Average B2 Glycoprotein IgM Titer (u/ml) | 35.5 (± 48.7) | 38.7 (± 46.8) | 0.725 |
| Anti Cardiolipin IgG positivity | 24 (92.3%) | 80 (51.2%) | <0.001 |
| Average Anti Cardiolipin IgG Titer (u/ml) | 138 (± 51.2) | 70.8 (± 68.3) | <0.001 |
| Anti Cardiolipin IgM positivity | 6 (23.1%) | 70 (44.9%) | 0.027 |
| Average Anti Cardiolipin IgM Titer (u/ml) | 25.9 (± 43.7) | 31.2 (± 39.9) | 0.207 |
| Lupus anticoagulant positivity | 25 (96.1%) | 114 (73.1%) | 0.028 |
| Triple Positive aPLs | 24 (92.3%) | 80 (51.2%) | <0.001 |
| Anti-nuclear Ab. (ANA) | 16 (61.5%) | 88 (56.4%) | 0.886 |
| Direct Coombs test positivity | 9 (34.6%) | 30 (19.2%) | 0.07 |
| Evans syndrome* | 6 (23.1%) | 10 (6.4%) | 0.007 |
| Severe thrombocytopenia (<50,000/µL) | 7 (26.9%) | 4 (2.5%) | <0.001 |
| C3 mg/dL (Average nadir) | 76.5 (CI95% 64.7–89.3) | 100.6 (CI95% 92.6–120.3) | 0.35 |
| C4 mg/dl (Average nadir) | 19.2 (CI95% 13.8–24.5) | 23 (CI95% 17.7–28.4) | 0.83 |
| Parameter | Terrible APS (n = 26) | APS control group (n = 156) | P value |
|---|---|---|---|
| Any microvascular = | 18 (69.2%) | 11 (7.1%) | <0.001 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| Heart Valve Disease | 16 (61.5%) | 19 (12.2%) | <0.001 |
|
|
|
|
|
|
|
|
|
|
| Livedo reticularis | 3 (11.5%) | 20 (12.8%) | 0.806 |
| Skin ulcer | 3 (11.5%) | 9 (5.7%) | 0.377 |
| Migraine | 3 (11.5%) | 28 (17.9%) | 0.377 |
| Epilepsy | 4 (15.3%) | 14 (9%) | 0.091 |
| Parameter | Terrible APS (n = 26) | APS control group (n = 156) | P value |
|---|---|---|---|
| Death of any cause | 5 (19.2%) | 8 (5.1%) | 0.012 |
| Death d/t APS event | 5 (19.2%) | 3 (1.9%) | <0.001 |
| Arterial thrombosis recurrence | 14 (53.8%) | 44 (28.2%) | 0.014 |
| Venous thrombosis recurrence | 15 (57.7%) | 23 (14.7%) | <0.01 |
| ≥2 breakthrough thrombotic recurrence * | 16 (61.5%) | 0 (0%) | <0.01 |
| >1 APS associated surgical intervention | 5 (19.2%) | 0 (0%) | <0.01 |
| Requirement for treatments with >1 immunomodulation ** | 9 (34.6%) | 0 (0%) | <0.01 |
| Any APS related surgical intervention# | 19 (73.1%) | 8 (5.1%) | <0.01 |
| Heart valve replacement | 7 (26.9%) | 3 (1.9%) | <0.01 |
| Vascular intervention (Thrombectomy Endarterectomy, Digit or limb amputation) | 13 (50%) | 5 (3.2%) | <0.01 |
| Obstetric morbidity recurrence | 3 (11.6%) | 39 (35.4%) | 0.07 |
| aGAPSS$ | 12.4 | 11.1 | 0.001 |
| aGAPSScvd$$ | 14.3 | 11.8 | 0.001 |
| Parameter | Terrible APS (n = 26) | Severe APS (n = 29) | P value |
|---|---|---|---|
| Age at presentation | 34 (CI95% 28.3–39.7) | 33.1 (CI95% 28.3–39.7) | 0.797 |
| Sex (% of females) | 19 (73.1%) | 19 (65.5%) | 0.83 |
| Hypertension | 6 (23.1%) | 9 (31%) | 0.517 |
| Smoker | 4 (15.3%) | 9 (27.5%) | 0.283 |
| Diabetes mellitus | 2 (7.6%) | 2 (6.9%) | 0.912 |
| Dyslipidemia | 6 (23.1) | 6 (20.6%) | 0.83 |
| Venous thrombosis at presentation | 15 (57.7%) | 18 (62.2%) | 0.946 |
| Arterial thrombosis at presentation | 16 (61.5%) | 11 (37.9%) | 0.04 |
| Triple Positive aPLs | 24 (92.3%) | 19 (65.5%) | 0.047 |
| Thrombocytopenia | 18 (69.2%) | 13 (44.8%) | 0.071 |
| Severe thrombocytopenia (<50,000/µL) | 7 (26.9%) | 3 (10.3%) | 0.11 |
| Any microvascular involvement | 18 (69.2%) | 6 (20.7%) | <0.001 |
| Heart Valve diseease | 16 (61.5%) | 9 (31%) | 0.023 |
| aGAPSS | 13.4 ( ± 2.39) | 11.7 ( ± 3.71) | 0.047 |
| Death of any cause | 5 (19.2%) | 3 (10.3%) | 0.409 |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsSystemic Lupus Erythematosus Research · Blood Coagulation and Thrombosis Mechanisms · Heparin-Induced Thrombocytopenia and Thrombosis
Introduction
Antiphospholipid syndrome (APS) is initially characterized by its association with thrombosis (e.g., stroke), obstetric complications, and the presence of lupus anticoagulant (LAC) (1, 2). Over time, this immune-mediated condition was further linked to a myriad of clinical manifestations, including macro- and microvascular thrombotic events, obstetric morbidity, heart valve disease, thrombocytopenia, and hemolytic anemia. Additionally, immunoassays used to detect antiphospholipid antibodies have evolved to include anti-cardiolipin (ACL) and anti-beta-2 glycoprotein (aB2GPI) autoantibodies alongside LAC (2–4). The classification criteria for APS have been revised several times and were established at the Sapporo summit 2006 (5) and recently redesigned in the new classification published in 2023 by the American College of Rheumatology and the European League Against Rheumatism (ACR-EULAR). The latter expanded the clinical criteria and refined the parameters for positive antibody assays (6). Furthermore, therapeutic guidelines published by EULAR emphasize the importance of homeostatic control in association with severity of disease as well as the role of immunomodulation with antimalarial drugs (7).
Despite improved understanding, definitions, and designed treatment protocols, clinicians still struggle, as the recurrence rate of APS manifestations ranges from 30% to 50% during long-term follow-up (8, 9). Moreover, APS disease progression is often unpredictable and may be severe or even catastrophic. Several decades ago, a very small subset of APS that manifests mainly with acute multiple microvascular thrombosis across different vascular beds within a short period, was defined as catastrophic APS (cAPS). This catastrophic variant is associated with high morbidity and mortality rates and requires intensive multifaceted interventions that have been found to be beneficial (10, 11). However, APS may present as a severe disease that is more gradual and does not fit the definition of cAPS. Severe APS appears to be more prevalent than cAPS and is less well-defined. In this line of thought, two APS severity scores were developed and validated: the adjusted Global Antiphospholipid Syndrome Score (aGAPSS) and the Damage Index for Thrombotic Antiphospholipid Syndrome (DIAP) (12–16). Additionally, some studies have investigated specific APS manifestations (i.e., heart valve disease or thrombocytopenia) as predictors of a worse disease course (17–19). Nonetheless, there is a lack of data on how to define severe and very severe APS, as well as the urgent need to improve disease outcomes, which potentially require a more aggressive approach and/or the addition of immunomodulation.
Hence, in this study we aim to identify a new variant of APS disease, which on the one side does not fit the cAPS definition yet on the other is extremely severe and refractory to current recommended therapy. We further aimed to understand what distinguishes this very severe variant and explore the possibility of developing a diagnostic tool to enable early detection of this variant, which may enable the study of tailored treatments for patients who present with this variant.
Objective
In this study, our objective was to define a specific subset of thrombotic antiphospholipid syndrome (APS) that is resistant, gradually progressive, and does not fit the criteria for catastrophic antiphospholipid syndrome (non-cAPS) (20). In addition, we aimed to identify key parameters associated with this very severe subgroup of APS and to develop a statistical tool that can assist clinicians in identifying patients at risk.
Methods
Patients
We enlisted all patients diagnosed with primary APS who were actively followed at our center between 2011 and 2024, and those who met the inclusion criteria for this study were included in the final analysis. Data were collected retrospectively to analyze demographic, clinical, and serological parameters, as well as details regarding disease progression. This study was performed in accordance with the Declaration of Helsinki and approved by the Sheba Medical Center Review Board.
Inclusion criteria
• Antiphospholipid syndrome (APS) was diagnosed according to the revised Sapporo Criteria (5).
• Primary APS patients—At Selection, all patients were diagnosed with primary antiphospholipid syndrome. A secondary autoimmune disease was documented if it developed during follow-up.
• Active regular follow-up (e.g., >1 follow-up visit within 12 months).
Exclusion criteria
• Catastrophic APS diagnosis according to the international consensus (10, 20).
• Non thrombotic disease (e.g., pure obstetric APS).
• Secondary APS at diagnosis.
• Non-criteria APS (e.g., not fulfilling the revised Sapporo APS criteria).
Clinical manifestations
We monitored APS-related events (e.g., thrombocytopenia and valve disease), specifically APS recurrences, including both thrombotic and non-thrombotic events. Notably, breakthrough thrombotic events were distinct from recurrent thrombotic events despite the use of full anticoagulant therapy (excluding DOAC therapy), absence of cardiovascular risk factors, and, in the case of venous thrombosis, absence of a provocation. Microvascular involvement was defined as the presence of one or more of the following: renal failure or proteinuria (with no other explanation), diffuse alveolar hemorrhage, or myocardial infarction without occlusive disease; a composite score of which any of the previous was positive, named any microvascular presentation. Microvascular involvement and other non-criteria manifestations were assessed at any time during follow-up.
We assessed the therapies required, including antiaggregation, anticoagulation, and immunomodulation, alongside any surgical interventions related to APS, mainly vascular procedures and heart valve repair or replacement. We also used the validated aGAPSS, which allots three points for dyslipidemia, one for arterial hypertension, five for anti-cardiolipin antibodies IgG/IgM, four for anti-β2 glycoprotein IgG/IgM, and four for lupus anticoagulant (12). We also calculated the proposed adjusted Global Antiphospholipid Syndrome Score for Cardiovascular Disease (aGAPSS-CVD), which incorporates factors such as smoking, diabetes, and obesity. This score emphasizes the risk of cardiovascular disease in patients with APS (21).
Serology
The presence of anti-cardiolipin (aCL) and anti-β_2_‐glycoprotein I (aβ_2_GPI) IgG and IgM isotypes was measured using enzyme‐linked immunosorbent assay (ELISA) or a multiplex system. The kits that were used were all commercial (ELISA—aB2GPI by AESKU Diagnostics (Wendelsheim, Germany) and aCL by Varelisa (Pharmacia Diagnostics, Stockholm, Sweden); Bioplex both aB2GPI and aCL (Bio-Rad, Hercules, CA, USA). B2GPI and ACL were considered positive if antibody levels were above 20 MPL units (IgG phospholipid units or IgM phospholipid units), or if >99th percentile or according to the manufacturer’s instructions were present in a minimum of two tests performed at least 12 weeks apart.
Lupus anticoagulant (LA) activity was detected using coagulation assays routinely performed at each center, consistent with the International Society of Thrombosis and Hemostasis guidelines (22). The LA assays were modified in 2016; until 2016, LA activity was measured by LA-responsive activated partial thromboplastin time (aPTT) aPL (by Stago, confirmed using the Actem FS Kit by Siemens, Erlangen, Germany), and from 2016, LA activity was measured by a combination of silicon clotting time and the use of the Russell Viper Venom Kit (IL, Bedford, MA, USA). In the case of anticoagulation treatment or spontaneous INR >1.5, patients’ plasma was mixed with normal plasma to reduce false positivity. Positivity was defined as single, double, or triple positivity according to the number of positive tests.
APS severity definition
In this study, three independent researchers reviewed all medical files and scored each patient’s disease as Mild, Intermediate, or Severe. If inconsistencies were documented between researchers, the patient file was re-reviewed in a joint meeting to reach a consensus. Notably, patients with Catastrophic APS (as per the definition of the CAPS registry group (10)) at any time during the disease were excluded. In addition, we compared patients with TrAPS to those with mild or intermediate disease.
Risk stratification:
A. Mild APS disease
No recurrence of APS manifestations.Thrombotic recurrence while treated with anti-aggregating therapy only, but no recurrence once switched to anticoagulation therapy.Recurrence due to cessation of therapy and/or poor adherence to therapy.No need for immunomodulation therapy other than hydroxychloroquine.
B. Intermediate APS disease (did not meet mild or severe criteria)
Thrombotic events post APS diagnosis, including any of the following:
- ◼ Transient ischemic attack with no lesions on magnetic resonance imaging.
- ◼ Arterial thrombotic events with frank cardiovascular risk factors (i.e., hypertension, dyslipidemia, smoking, or diabetes mellitus).
- ◼ Venous recurrence while patients on low dose anti-coagulation therapy (e.g., low molecular weight heparin 0.5 mg/kg once daily).
- ◼ Thrombotic recurrence (either arterial or venous) while treated with direct oral anticoagulants.
- ◼ Use of immunomodulation with low-dose glucocorticoids and/or cDMARDs (e.g., azathioprine) only.
- ◼ Surgical interventions due to APS manifestations while non-adherence to anticoagulation therapy.
Severe APS disease (at least one of the following):
≥1 Breakthrough Events
- ○ or Surgical intervention (e.g., need for limb amputation) due to APS despite treatments according to the current guidelines.
- ○ or APS manifestations that required treatment with one advanced immunomodulation (i.e., rituximab, intravenous immunoglobulin, or plasma exchange therapy).
Terrible APS (TrAPS) variant was defined if:
2 Breakthrough Events
- or
1 surgical intervention (e.g., pulmonary endarterectomy, heart valve surgery, and/or limb amputation, etc.).
- ○ or APS manifestation that required treatment with >1 with advanced immunomodulation (i.e., combination of rituximab, intravenous immunoglobulin, or plasma exchange therapy, etc.).
Statistical analysis
The data were analyzed using jamovi version 2.6 [Computer Software], developed by The Jamovi Project (2025). This can be accessed at https://www.jamovi.org. To analyze between-group differences in discrete variables, either Pearson’s chi-square test or Fisher’s exact test (two-tailed) was employed. Variables that were found to be significant (p <0.10) in the univariate analysis were subsequently used in a stepwise logistic regression to identify those most significantly associated with each outcome measure. The selection of variables for the stepwise analysis was limited based on the total number of patients, allowing for one variable for every five patients in the study. Patients with missing data were excluded from the analysis. Odds ratios (ORs) and 95% confidence intervals (CIs) were calculated, with a significance level set at p ≤0.05.
Results
In this study, 306 patients with primary APS were assessed, and upon review and exclusion based on our study criteria, 209 patients with thrombotic APS were designated as suitable for severity stratification. Of these, study group included 27 of 209 (12.7%) patients with the new variant TrAPS, compared to a control group of 156 patients, a composite of mild and moderate APS disease (Figure 1).
Our cohort of APS patients. Bold—Analyzed cohort, Italic—control group; Underlined—Study group.
Demography, cardiovascular risk factors, and APS presenting manifestations of TrAPS vs. APS controls
Our study and control groups were similar in terms of age, sex, and cardiovascular risk factors (Table 1). The rate of additional autoimmune disease diagnosis during follow-up was low (5%–10%) and did not differ between the groups. Notably, the follow-up time among patients with TrAPS was longer, with an average of 4.9 years. Additionally, venous thrombosis at presentation was associated with the diagnosis of TrAPS during follow-up. Obstetric morbidity at presentation was inversely associated with TrAPS, although this difference did not reach statistical significance. Interestingly, in the TrAPS group, five patients upon presentation had signs of both venous thrombosis and arterial thrombosis (ex. signs of pulmonary embolism on CTA in the stroke protocol or signs of ischemic events on brain imaging during patient evaluation for pulmonary embolism), this patient did not fulfil the criteria for CAPS.
Laboratory findings and serology
The diagnosis of TrAPS was linked to the presence of triple aPL positivity, which was mostly evident for anti-β2-Glycoprotein I and anti-cardiolipin of the IgG subtypes. Interestingly, anti-cardiolipin antibodies of the IgM isotype (ACL IgM) were inversely correlated with TrAPS. In addition, APS-associated thrombocytopenia of any severity was linked to TrAPS (data not shown), while extreme thrombocytopenia (<50,000) was highly prevalent in our study group, as well as clinical evidence of both hemolytic anemia and thrombocytopenia, defined as Evans syndrome (Table 2).
Microvascular, heart valve involvement, and non-criteria manifestation
Nephropathy, diffuse alveolar hemorrhage, and myocardial disease were significantly more common in the TrAPS group, whereas livedo reticularis and other non-criteria manifestations (skin ulcers, migraine, and epilepsy) did not differ from those in the control group (Table 3).
APS disease outcomes and treatments
Mortality was almost triple in the TrAPS group, which was statistically significant (Table 4). Focusing on the causes of death in the control group, three individuals died from malignancies, three from complications related to cerebrovascular accidents (CVA), one from COVID-19, and one from sepsis. In contrast, all causes of death in the TRAPS group were linked to APS morbidity rather than other disease processes, reflecting the aggressive and refractory course of this condition.
Given the inclusion criteria and research methodology, it is not surprising that the rate of thrombotic recurrence was higher in the TrAPS group. Even milder forms of APS exhibit a significant recurrence rate despite anticoagulant treatment, with more than 80% of patients within the TrAPS group experiencing this type of recurrence (Table 4). Particularly, breakthrough events—defined as thrombosis despite full anticoagulant therapy (DOAC therapy excluded), with no other cardiovascular risk factors—were present only in the TrAPS group (per design), as 18/27 (66%) patients had two or more such events (Table 4). Nonetheless, both aGAPSS and aGAPSS-CVD scores were significantly higher in the TRAPS group, confirming their validity as tools for assessing the severity of antiphospholipid syndrome (APS).
In this study, treatments, either surgical or medical, were according to the treated physician dissertation. The TrAPS group was particularly resistant to anticoagulant therapy, and 81.4% (22 of 27 patients) of patients with TrAPS required significant adjunctive therapy. Of which 2/3 of patients with TrAPS required multiple types of immune modulation (i.e., two or more of IVIG, rituximab, or plasmapheresis) to achieve disease control. Moreover, 19/27 (70.3%) patients with TrAPS required surgical intervention, while 18.5% (5/27) required multiple surgical interventions (Table 4).
Multivariate analysis and TrAPScore design
In our univariate analysis, we found that TrAPS was associated with venous thrombosis at presentation, longer follow-up, microvascular manifestations, triple aPLA positivity (particular IgG subtypes, both in positivity and titter, as well as the presence of lupus anticoagulant), thrombocytopenia, and Evans syndrome. In comparison, obstetric morbidity at presentation and IgM aCL antibodies were associated with less severe disease.
To better understand which parameters correlated most with the TrAPS group, we performed a multivariate analysis, in which four parameters proved to be the most significant (area under the curve, 0.902). Based on this ratio, we developed the TrAPScore (Figure 2).
Visual figure of the TrAPScore.
Severe thrombocytopenia OR 9.7 (CI95% 1.55–60.8) TrAPScore 4 points.Heart valve disease OR 8.8 (CI95% 2.9–26) TrAPScore 4 points.Microvascular involvement OR 7.1 (CI95% 2–21) TrAPScore 3 points.Triple positive aPLs OR 5.5 (CI95%1.4–22.9) TrAPScore 2 points.
To provide cutoffs for the TrAPScore that may enable the prediction of TrAPS, the TrAPScore was evaluated across different comparisons. When distinguishing the TrAPS group (n = 27) from the APS control group (n = 156) using a cutoff score >6, the test demonstrated a sensitivity of 51.4% and a high specificity of 97.4%. The positive predictive value (PPV) was 77.8% and the negative predictive value (NPV) was 92.1%, with an overall accuracy of 90.7%. Based on this analysis, we concluded that TrAPScore >6 is a plausible cutoff for defining TrAPS.
To establish an appropriate cutoff for a negative TrAPScore, we determined that a TrAPScore <4 effectively differentiated the TrAPS group from the APS control group. At this threshold, the sensitivity was 85.2%, and the specificity was 79.5%. The positive predictive value (PPV) was 41.8%, whereas the negative predictive value (NPV) was notably high at 96.9%. The overall accuracy of this cutoff was 80.3%. A TrAPScore of less than 4 thus demonstrates a high NPV of 96.9%, suggesting that it could serve as a reliable exclusion criterion for diagnosing this APS variant (Figure 3).
TrAPS score mild/moderate and TrAPS.
TrAPS and severe APS
Based on the study design, we initially compared TrAPS with mild or moderate disease. To better understand whether TrAPS is distinguishable from other forms of APS, we compared the clinical features of severe APS (Table 5). The groups exhibited less pronounced differences, with notable variations primarily observed in the components of the TrAPS score, namely, heart valve disease, microvascular involvement, and triple positivity. Although severe thrombocytopenia was more common in the TrAPS group, this difference was not statistically significant. Additionally, mortality rates were nearly twice as high in the TrAPS cohort; however, this difference was not statistically significant.
In the comparison between the TrAPS group (n = 27) and the severe APS group (non-TrAPS; n = 28), the same cutoff yielded a sensitivity of 51.8%, specificity of 88%, PPV of 82.5%, NPV of 63.5%, and overall accuracy of 69% (Figure 4).
TrAPScore in severe APS vs TrAPS.
Discussion
In this study, we aimed to provide a new perspective on APS severity by defining a novel variant with a particular focus on long-term resistance to recommended therapies. This aim arises from the patient’s bedside, as resistant APS is not an infrequent challenge. Notably, TrAPS differs from catastrophic-APS in terms of both the time frame and pattern of manifestations. Nonetheless, TrAPS like cAPS, may require a more aggressive approach to control.
The notion of better defining APS severity and/or APS risk stratification stands in agreement with several previous attempts that resulted in compelling and well-validated scores (13, 15). One might wonder why another risk stratification is needed, underscoring the focus of TrAPS inception to address the unmet need to define a new clinical tool for therapy-resistant disease. In this line of thought, several recent publications have focused on the use of immune modulating agents, particularly in resistant APS (23–25). Others have tried to focus on the clinical aspects that would be resistant to anticoagulant therapy (26). A recent review of new treatment targets deemed immunomodulatory therapies to be reserved for those with refractory disease and microvascular and non-thrombotic manifestations of APS (27). The goal of this study was to address and this unmet need.
Hence, we selected a homogeneous group of primary thrombotic APS patients, excluding secondary APS, non-thrombotic patients (i.e., OAPS), and non-criteria diseases. All patients were under active follow-up by a dedicated team in our APS center, which enabled a thorough assessment of disease progression, compliance with treatment, and evaluation of treatment outcomes. We applied specific objective criteria, including the definition of breakthrough thrombotic events despite full anticoagulant therapy for arterial thrombosis, the absence of other cardiovascular risk factors, and the lack of appropriate provocation for venous thrombosis, all of which have been detailed in the new classification criteria for APS (6). We also considered other markers of resistance to therapy, such as the need for immune modulation (such as anti-CD20 therapy, IVIG, or plasma exchange) and/or surgical interventions (e.g., heart valve replacement or vascular surgery) related to APS.
In this cohort, 27/209 (12.7%) of the thrombotic APS patients were included in the TrAPS group. This unique group had a specific profile at presentation, which was associated with venous thrombosis, higher aGAPSS score, and the presence of aPLs of the IgG subtype. In contrast, obstetric morbidity and aPLs of the IgM subtype may be considered “protective,” suggesting an interesting role for different aPLs in APS pathogenesis, which remains to be elucidated. As presumed outcomes of the TrAPS group were worse, mortality was higher (18.5% vs. 5.1%, p = 0.012), particularly due to APS events (18.5% vs. 1.9%, p <0.001) compared to the APS control, as well as the need for immune modulation or surgery.
Finally, we aimed to develop a tool that would assist clinicians in predicting TrAPS using the TrAPScore. The latter was based on a multivariate regression analysis using four parameters, each graded in regard to its relative impact: severe thrombocytopenia (4 points), heart valve disease (4 points), microvascular involvement (3 points), and triple-positive serology (2 points). Utilizing this score, values higher than 6 resulted in a positive predictive value of 78%–82.5%, while a value lower than 4 had a negative predictive value of 96.5%. Although this score requires further validation through additional studies, we believe it has the potential to provide a quantitative measure of a relatively common unmet clinical need. Whether this tool will effectively assist clinicians in navigating therapy for this challenging subset of patients remains to be determined.
The strength of both TrAPS and the TrAPScore is most evident when comparing this group to severe APS patients—the latter defined as those who experienced no more than one breakthrough event, required sporadic immunomodulation, or APS-related surgery but overall maintained disease control. In this context, differences in clinical presentation and antibody titers were less pronounced. Additionally, the commonly used severity index, aGAPSS, showed some differences; however, these distinctions may not be sufficient to definitively classify patients as having a truly refractory disease. As illustrated in the figure, the TrAPScore demonstrates a clear separation between patients with severe APS and those with TrAPS, highlighting its potential utility in distinguishing clinically untreatable cases.
This study is not free of limitations derived from its retrospective single-center design regarding patient selection and disease severity estimation. However, this approach enabled us to assess severity based on long-term follow-ups, and we applied three independent re-assessments of the clinical course for each patient to better define the risk factors. Second, the institution of a new severity subgroup is somewhat subjective in deciding what qualifies as severe, which requires further validation in different cohorts. Lastly, the fact that our center is a tertiary referral center might bias our APS cohort as patients with more severe disease are typically referred; this might be reflected by the relatively large group of cAPS in our cohort [4.6% vs. approximately 1% in previous reports (11)]. We believe that these biases are addressed with the design of this study, that is, objective criteria in patient selection, a homogeneous study group (primary thrombotic APS patients), active follow-up/monitoring of all patients, and multivariate statistical analysis. Nevertheless, this finding requires confirmation in future studies with different cohorts. Nonetheless, we hope that these findings will address a clinical need and be used by other clinicians to evaluate the accuracy of our findings.
Conclusion
This study evaluated a new subset of resistant thrombotic APS disease, termed “terrible APS”—TrAPS. This new variant of APS was defined by the presence of multiple breakthrough thrombotic events despite proper anticoagulation and/or the need for immune-mediated treatments and/or requirement for APS-associated surgical intervention.
The TrAPScore was used to assess this variant of the disease based on four parameters: severe thrombocytopenia (4 points), heart valve disease (4 points), microvascular involvement (3 points), and triple positive aPLs (2 points). Based on our data, a TrAPScore higher than 6 had a positive predictive value of 78%–82.5%, whereas a value lower than 4 had a negative predictive value of 96.5%. Future studies are needed to determine whether this score truly differentiates cases that are resistant to most current treatments and will require reassessment of the management of these cases.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Hughes GR . Thrombosis, abortion, cerebral disease, and the lupus anticoagulant. Br Med J Clin Res Ed. (1983) 287:1088–9. doi: 10.1136/bmj.287.6399.1088, PMID: 6414579 PMC 1549319 · doi ↗ · pubmed ↗
- 2Schreiber K Sciascia S de Groot PG Devreese K Jacobsen S Ruiz-Irastorza G . Antiphospholipid syndrome. Nat Rev Dis Primer. (2018) 4:1–20. doi: 10.1038/nrdp.2017.103, PMID: 29368699 · doi ↗ · pubmed ↗
- 3Ruiz-Irastorza G Crowther M Branch W Khamashta MA . Antiphospholipid syndrome. Lancet. (2010) 376:1498–509. doi: 10.1016/S 0140-6736(10)60709-X, PMID: 20822807 · doi ↗ · pubmed ↗
- 4Knight JS Branch DW Ortel TL . Antiphospholipid syndrome: advances in diagnosis, pathogenesis, and management. BMJ. (2023) 380., PMID: 36849186 10.1136/bmj-2021-069717 · doi ↗ · pubmed ↗
- 5Miyakis S Lockshin MD Atsumi T Branch DW Brey RL Cervera R . International consensus statement on an update of the classification criteria for definite antiphospholipid syndrome (APS). J Thromb Haemost. (2006) 4:295–306. doi: 10.1111/j.1538-7836.2006.01753.x, PMID: 16420554 · doi ↗ · pubmed ↗
- 6Barbhaiya M Zuily S Naden R Hendry A Manneville F Amigo MC . The 2023 ACR/EULAR antiphospholipid syndrome classification criteria. Arthritis Rheumatol. (2023) 75:1687–702. doi: 10.1002/art.42624, PMID: 37635643 · doi ↗ · pubmed ↗
- 7Tektonidou MG Andreoli L Limper M Amoura Z Cervera R Costedoat-Chalumeau N . EULAR recommendations for the management of antiphospholipid syndrome in adults. Ann Rheum Dis. (2019) 78:1296–304. 10.1136/annrheumdis-2019-215213 PMC 1103481731092409 · doi ↗ · pubmed ↗
- 8Bazzan M Vaccarino A Stella S Sciascia S Montaruli B Bertero MT . Patients with antiphosholipid syndrome and thrombotic recurrences: A real world observation (the Piedmont cohort study). Lupus. (2016) 25:479–85. doi: 10.1177/0961203315617538, PMID: 26657216 · doi ↗ · pubmed ↗