Synthesis and meta-Amination of Pyridines via Multicomponent Cascade Reaction
Telmo N. Francisco, Nuno R. Candeias, Samuel Guieu, Joana L. C. Sousa, Rafael F. A. Gomes, Carlos A. M. Afonso, Artur M. S. Silva, Hélio M. T. Albuquerque

TL;DR
This paper introduces a new green method to synthesize 3-aminopyridines using a cascade reaction, which can be adapted to make various substituted pyridines.
Contribution
A novel multicomponent cascade synthesis of 3-aminopyridines via Zincke aminolysis with ammonia is presented.
Findings
The method shows broad scope with over 20 examples and good functional group tolerance.
It allows scalable synthesis in both batch and flow procedures.
The approach enables the production of 3-halo, 3-hydroxy, and 3-thiopyridines through peripheral editing.
Abstract
We report a green, multicomponent cascade synthesis of 3-aminopyridines via [1,3′-bipyridin]-1-ium intermediates undergoing Zincke aminolysis with ammonia supported by experimental evidence and DFT calculations. The method offers broad scope (>20 examples), good functional group tolerance, and scalability in batch and flow procedures. Synthetic utility is demonstrated through conversion into 3-halo, 3-hydroxy, and 3-thiopyridines, expanding the access to meta-substituted pyridines through peripheral editing of the unprotected amino group.
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
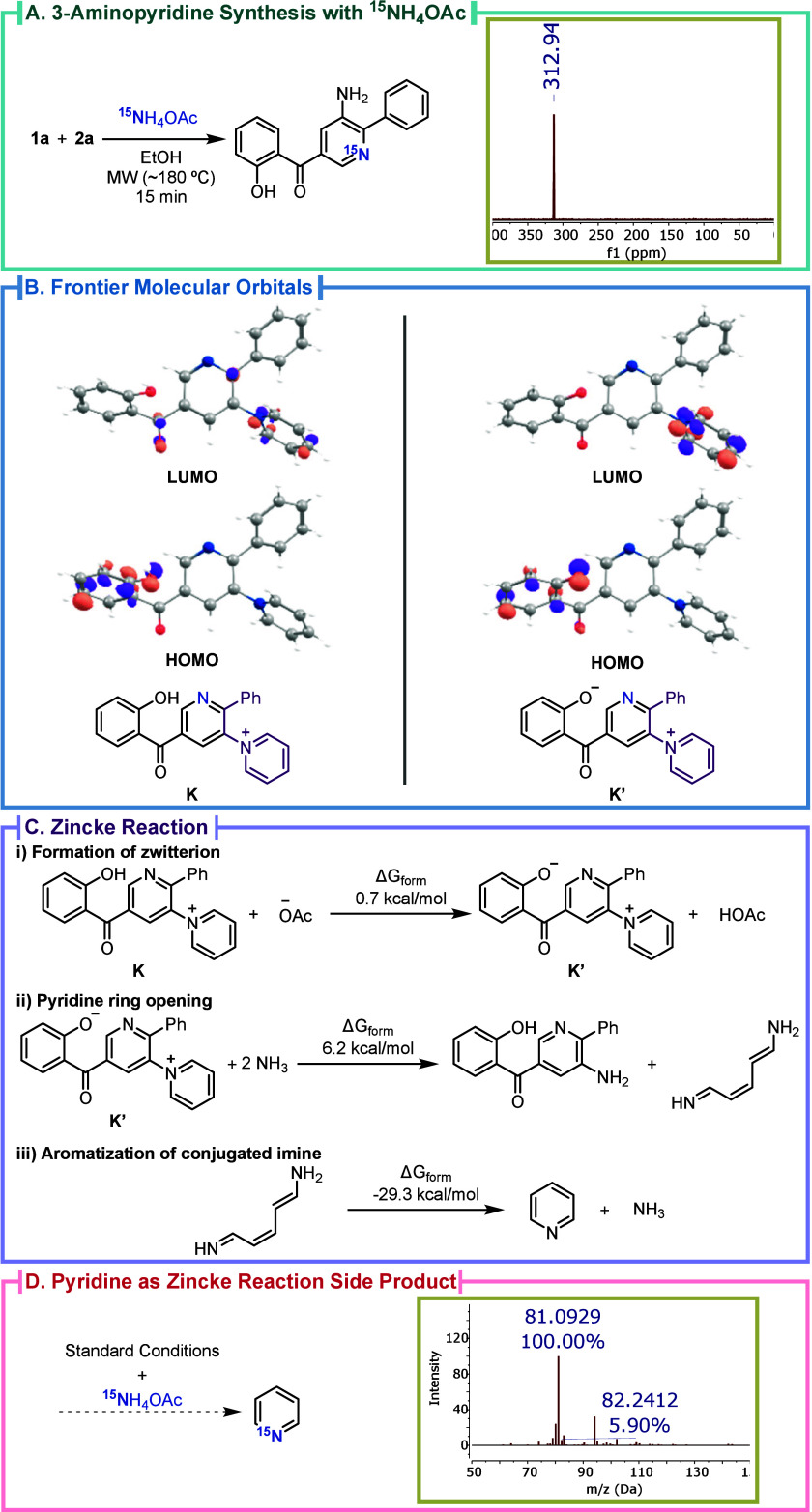 Figure 1
Figure 1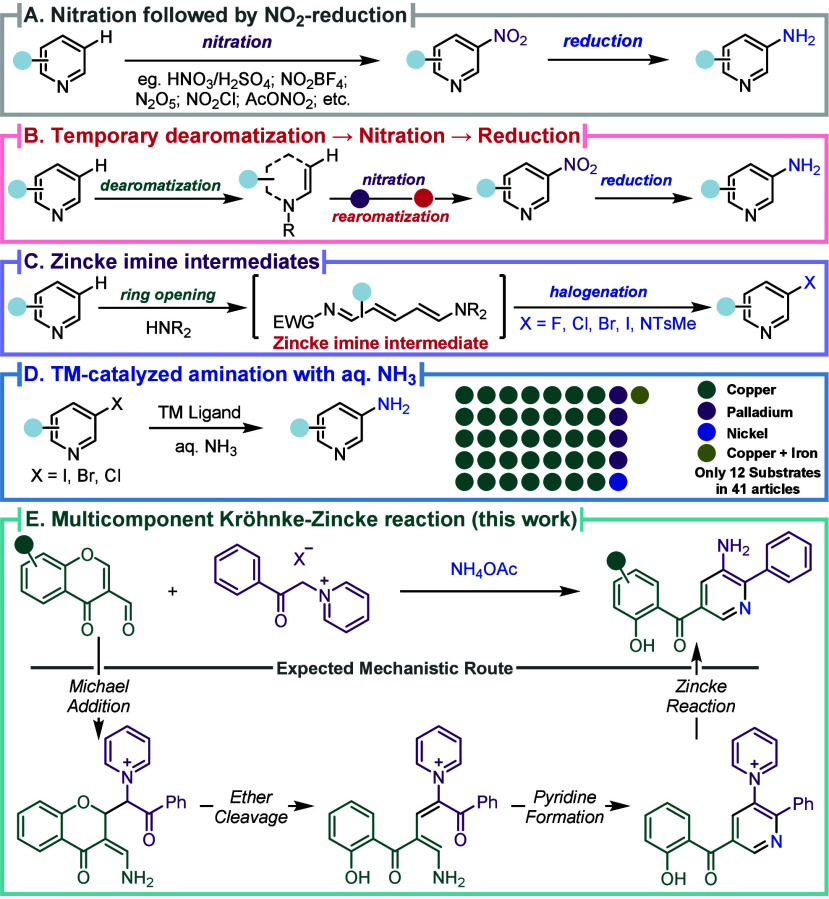 Figure 2
Figure 2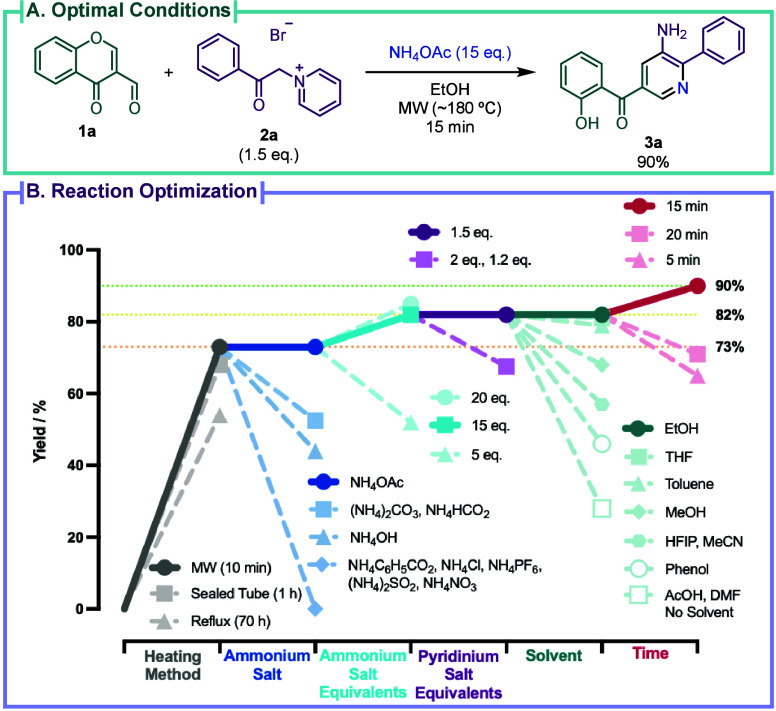 Figure 3
Figure 3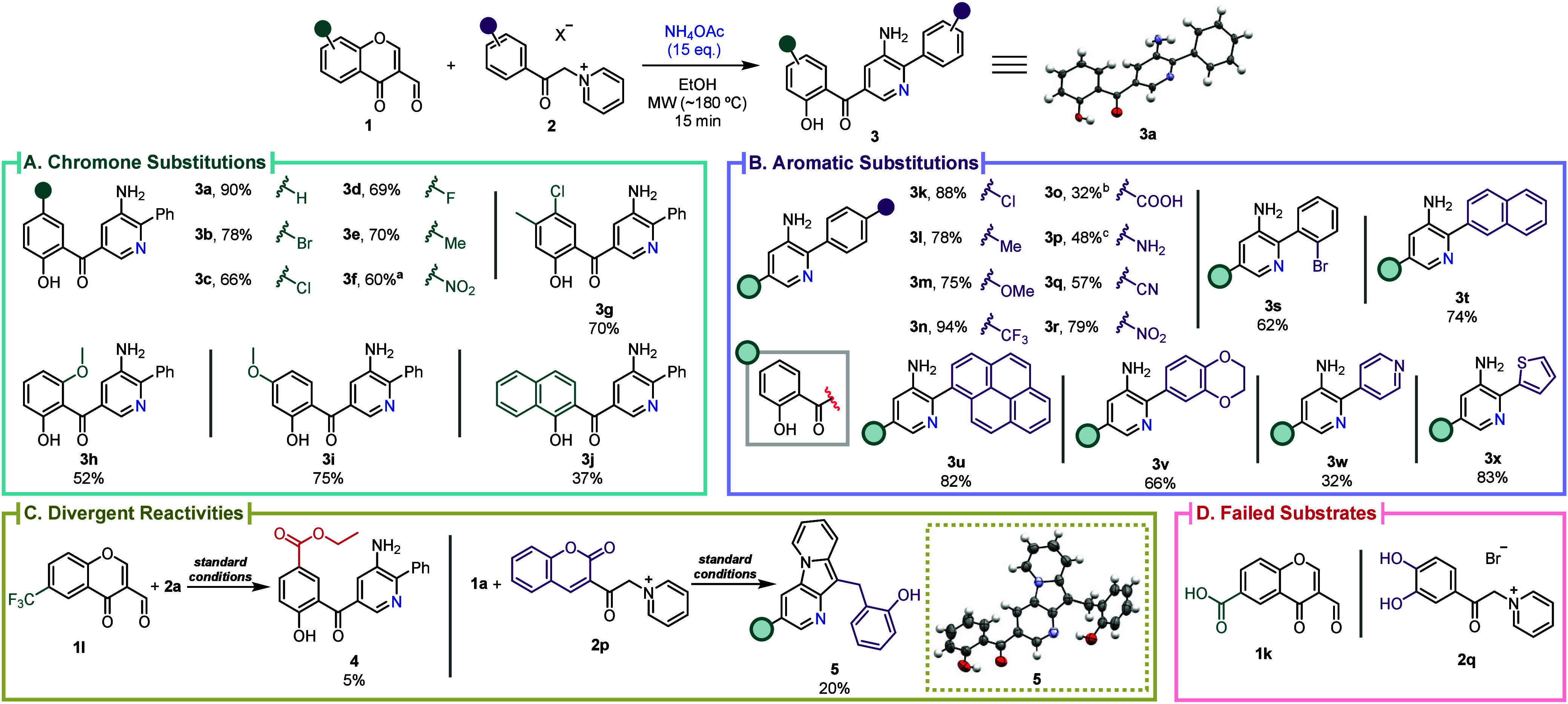 Figure 4
Figure 4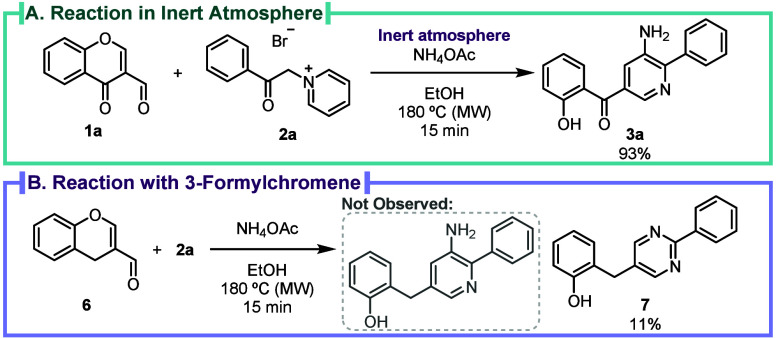 Figure 5
Figure 5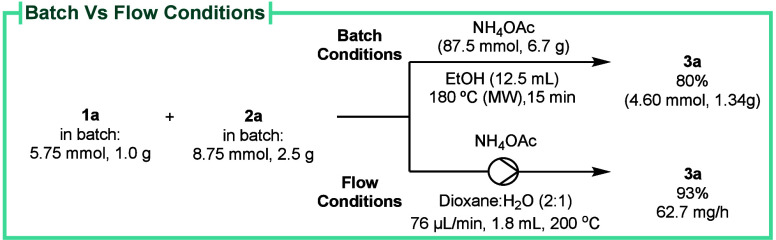 Figure 6
Figure 6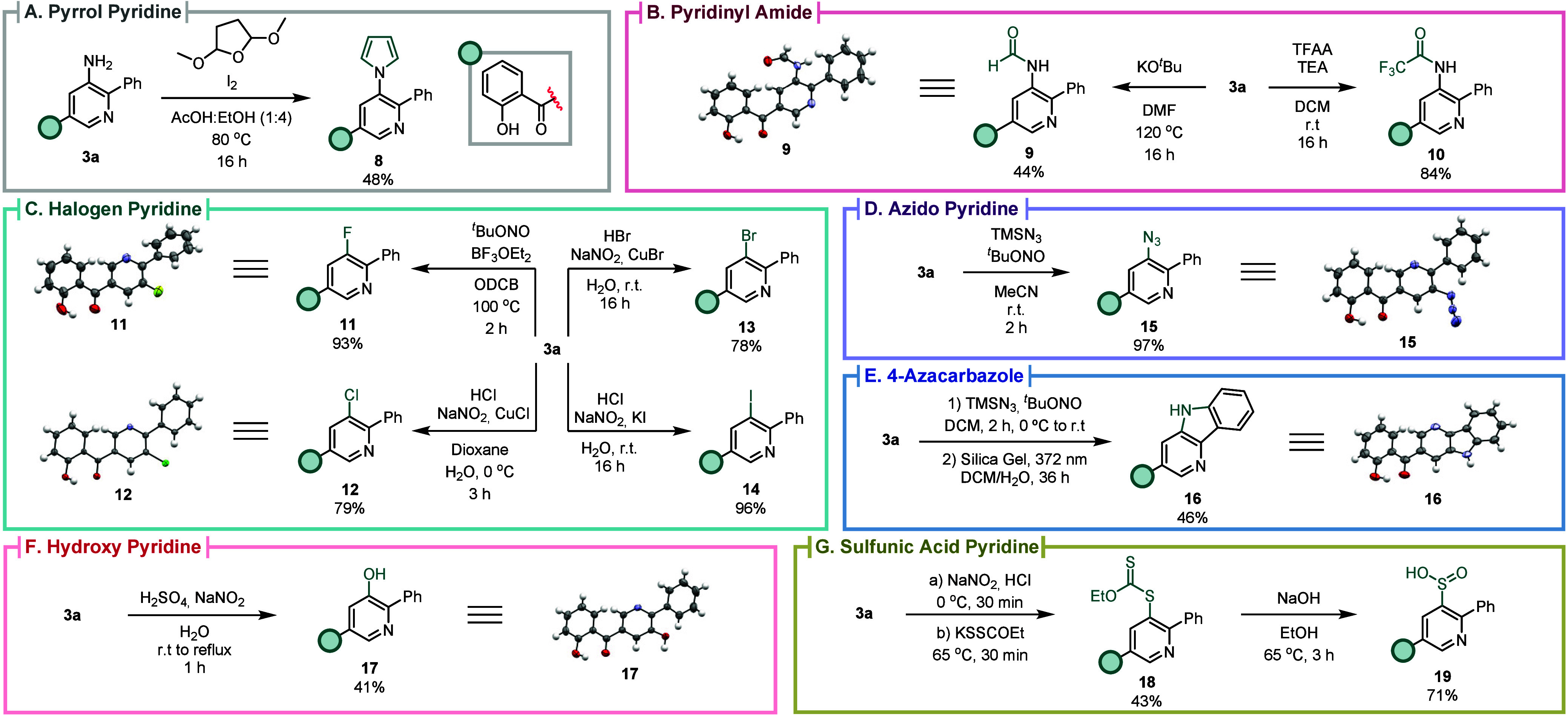 Figure 7
Figure 7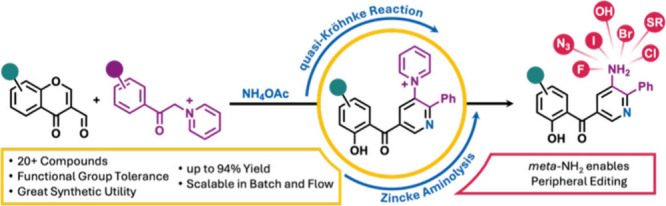 Figure 8
Figure 8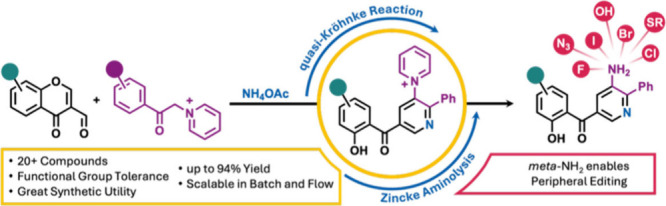 Figure 9
Figure 9Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsCatalytic C–H Functionalization Methods · Phenothiazines and Benzothiazines Synthesis and Activities · Chemical Synthesis and Analysis
Pyridines are crucial in biological, material, and agrochemical sciences, ?−? ? ? topping the list of heterocycles in FDA-approved drugs,? standing in 18% of the top-selling agrochemicals,? and appearing as building blocks in ligands and functional materials. ?−? ? The electronic properties of pyridines hinder meta-substituted derivative preparation compared to ortho- and para-substituted isomers. ?,? Literature analysis (Figure S1) revealed that approximately 99% of the transformations for the synthesis of meta-aminopyridines already had a preconstructed pyridine ring, with a nitrogen-containing precursor in the meta-position. Around 89% of synthetic pathways involve nitro group reduction protocols preceded by nitration steps, usually requiring harsh conditions leading to low levels of chemo and regioselectivity for the meta-position (SchemeA). ?,? Other strategies based on nitrogen-containing precursors are also limited by low yields and use of specific and of limited availability reagents, with limited substrate scope. ?−? ? ? ? Modern approaches have been reported for meta-selective pyridine C–H functionalization relying on the use of directing groups, nondirected metalation, or temporary dearomatization. ?,?−? ? ? Important developments have been made by Studer regarding the meta-fluorination, hydroxylation, and nitration of pyridines upon temporary dearomatization strategy (SchemeB). ?−? ? ? Another elegant approach for the meta-selective pyridine halogenation involving Zincke imine intermediates was recently developed (SchemeC). ?−? ? However, these methods fail for direct meta-selective amination of pyridines with unprotected amino groups. Although C3-aminated pyridines with the NTsMe group have been achieved, this method has regioselectivity issues and cannot produce pyridines with C3 unprotected amino groups even after further transformations and using expensive iridium catalyst (SchemeC).? An interesting option in this regard is the use of pyridine (pseudo)halides through transition-metal catalytic amination. ?−? ? Nonetheless, this strategy is limited to tertiary and secondary amines, whereas the primary ones are extremely rare and underdeveloped. ?,?,? For the latter case, the use of Pd- or Cu-catalyzed procedures with aqueous ammonia contributed to this challenge (SchemeD), although very few examples of 3-aminopyridines have been produced and very careful ligand control must be considered. ?,? Despite these advances, straightforward access to pyridines functionalized with the “free” amino group (−NH_2_) at the meta-position (C-3) remains undiscovered.
Herein, we report a multicomponent reaction (MCR) platform for the one-pot assembly of 3-aminopyridines from readily accessible materials, such as 3-formylchromones, pyridinium, and ammonium salts (SchemeE). The ammonium salt plays a double role in the reaction mechanism, being involved in the construction of the pyridine moiety and as the initiator of the Zincke reaction. The key feature of this cascade reaction is that the chromone ether-cleavage enables the aromatization of the pyridine ring without removal of the pyridinium, granting a subsequent Zincke reaction to complete the synthesis of 3-aminopyridines.
The initial experiments to efficiently deploy this MCR involved extensive optimization (SchemeC, Tables S1–S3) using 3-formylchromone 1a and pyridinium salt 2a as substrates, synthesizing 3-aminopyridine 3a in 90% yield with the optimal conditions (SchemeA), as confirmed through crystal X-ray diffraction (SchemeB). Preliminary studies demonstrated that the reaction was more effective under microwave (MW) irradiation, although it was not essential for obtaining 3-aminopyridine 3a. NH_4_OAc proved to be the best nitrogen source, with (NH_4_)2_CO_3 and NH_4_HCO_2_ as suitable alternatives. The MCR demonstrated compatibility with a wide range of protic and nonprotic solvents, including H_2_O, with EtOH, THF, and toluene as the best options. It is noteworthy that MeOH exhibited comparable results to the reaction using EtOH; however, the formation of imine S2 (Figure S3) as a byproduct resulted in lower yields.
Having set the optimal reaction conditions, we investigated the scope of this MCR platform by evaluating various 3- formylchromones 1 and pyridinium salts 2 (Scheme). The methodology was compatible with electron-donating (EDGs), electron-neutral, and electron-withdrawing groups (EWGs) at different positions of chromone components 1a–j, with the corresponding 3-aminopyridines 3a–j obtained in 37–90% yield (SchemeA). Even sterically encumbered substrates proved successful, despite the lower yield for 3j. Notwithstanding, this methodology was incompatible with two chromone substrates, 1k and 1l (SchemeC). The presence of a COOH group in the 3-formylchromone 1k, led to the formation of the decarboxylated 3-aminopyridine 3a. The presence of a CF_3_ group as substituent, 1l, did not produce the expected 3-aminopyridine, even with THF or toluene; instead, amidst multiple side reactions and/or decomposition, the 3-aminopyridne 4 was isolated in 5% yield, resulting from the reaction with EtOH under alkaline conditions after the CF_3_ group was anionically activated (SchemeC).? Following the scope studies, we surveyed the viable substitutions that could be applied to pyridinium salts 2, leading to the synthesis of 3-aminopyridines 3k–s in 32–94% yield (SchemeB). The methodology was well-suited for EDGs, neutral, and EWGs, tolerating the presence of sensitive COOH (3o), CN (3q), and NH_2_ (3p) groups. Sterically demanding groups such as naphthalene (3t) and pyrene (3u) are better tolerated in pyridinium salts than in 3-formylchromone partners. The reaction was also compatible with O-, N-, and S-heterocycles such as dioxane, pyridine, and thiophene delivering pyridines 3v–x in 32–83% yield (SchemeB). When using the pyridinium salt 2p, the coumarin moiety would engage a divergent reactivity at the ortho-position of the pyridinium leading to the formation of pyrido indolizine 5 (SchemeC). The presence of the COOH moiety resulted in the formation of the corresponding ester S4 (Figures S96 and S97) through reaction with EtOH, which could be hampered by replacing the EtOH by THF. Similarly, when synthesizing 3-aminopyridine 3p, (NH_4_)2_CO_3 was used as the nitrogen source, to avoid formation of amide S5 (Figures S98 and S99) observed when NH_4_OAc was used. The presence of OH groups in the aromatic ring (2q) was incompatible with this procedure due to side reactions and/or decomposition (SchemeC).
To elucidate the mechanism, density functional theory (DFT) calculations and experimental studies were conducted. Considering the formylchromone imine as an energetically favorable intermediate for Michael addition and the ylide as the starting set of reactants (see Scheme S1 for further details), the pyridine moiety construction mechanism has two principal phases: the addition of ylide to chromone imine and the cyclization phase where the key [1,3′-bipyridin]-1-ium intermediate K (detected with HPLC-MS) is formed (Figures S11 and S16). From here on, the electronically biased pyridine by the presence of the pyridinium moiety in the intermediate K could lead to the substitution of the pyridinium with ammonia, or the Zincke reaction to liberate the 3-NH_2_ group. The control reaction employing ^15^NH_4_OAc (FiguresA and S12–S15) revealed that the ammonia identified in the reaction mixture did not serve as the source of the 3-amino group. Therefore, the amino group is derived from the pyridinium moiety after Zincke aminolysis with ammonia. The presence of a strong electron acceptor such as the dinitrophenyl (DNP) group is usually an electronic requirement for a Zincke reaction to take place. ?,? Nevertheless, such a transformation would originate the herein-described 3-aminopyridines. The analysis of the frontier orbitals of cation K and its zwitterion analogue K′ shows a clear difference in the location and coefficient of the LUMO orbitals (FigureB).Conversion of cation K to zwitterion K′ is slightly endergonic but becomes exergonic at higher theory levels. Ring opening to 3-aminopyridine is moderately uphill, whereas pyridine formation via rearrangement or intramolecular addition is strongly favored, with aromatization driving the process (FigureC). The analysis of HPLC-MS of the crude of the 3-aminopyridine synthesis using ^15^N-labbeled ammonium acetate (Figure S17) allowed the detection of the ^15^N-pyridine which further supports the Zincke reaction hypothesis (FigureD). Two control reactions were performed to support the reaction mechanism. The reaction was carried out under an inert atmosphere (SchemeA) to determine whether the presence of air could be key in the aromatization of the pyridine. The synthesis of 3-aminopyridine 3a was successfully accomplished under these conditions, demonstrating a marginal increase in yield (90% in air and 93% in an inert atmosphere), indicating that air is not essential for the aromatization step. Next, 3-formylchromone 1a was switched to 3-formylchromene 6 (SchemeB) to verify the effect of the ketone moiety in the reaction. Pyrimidine 7 (see Scheme S5 for mechanistic details) was obtained instead of the expected 3-aminopyridine, implying that the carbonyl was a key element, according to the DFT studies, in the stabilization of intermediate B after the conjugate addition of the ylide to the chromone imine.
One-pot, scalable, and metal-free protocols for the synthesis of amino group-containing pyridines are highly desirable, as drug molecules typically contain amines and N-heterocycles.? The scalability of the reaction was first studied by reproducing the reaction under MW irradiation at gram-scale with a slight yield decrease (Scheme). Therefore, to convey a more appropriate option for the large-scale production of 3-aminopyridines, we performed preliminary studies to adapt this methodology to continuous flow conditions. After optimization (monitored by HPLC), by using a mixture of dioxane and water (2:1) and a packed bed reactor using sand as the solid material (see SI for details), 3-aminopyridine 3a was produced in 81% yield. We also verified that, at the stationary state, 3-aminopyridine 3a has been produced in 95–100% yield. Following a duration of slightly above 5 h, 327.3 mg of 3-aminopyridine 3a was obtained in 93% yield after isolation (Scheme).
The NH_2_ group in 3a serves as a versatile synthetic handle that can be efficiently transformed into various functional groups (Schemes): for instance, the conversion into a pyrrole for the synthesis of pyridine 8 in 48% yield (SchemeA), and the conversion into amides 9 and 10 in 44% and 84% yield, respectively (SchemeB). Subsequently, the amino group of 3a was used to install a wide array of substituents in the meta-position, such as halogens (F, Cl, Br, and I) leading to pyridines 11, 12, 13, and 14 in 78–96% yield, through the respective diazonium salts (SchemeC). It was also possible to install an azide group to prepare pyridine 15 in 97% yield (SchemeD), as well as the cyclization of 3a into 4-azacarbazole 16 in 46% yield (SchemeE). The 3-aminopyridine 3a can be converted into 3-hydroxypyridine 17 in 41% yield (SchemeF), while treatment with potassium ethyl xanthate led to the pyridinyl carbonodithioate 18 in 43% yield, which was then converted into pyridine-3-sulfinic acid 19 in 71% yield upon treatment with NaOH (SchemeG).
In summary, we have developed a new methodology for the synthesis of challenging 3-aminopyridines through a multicomponent cascade reaction characterized by a broad substrate scope, easy operation, and scalability in both batch and flow. The 3-aminopyridines revealed compelling synthetic utility and were subsequently converted into multiple meta-functionalized pyridines of high interest. Subsequent investigations to deploy this MCR platform in a diversity-oriented synthesis (DOS) context are underway in our laboratory.
Supplementary Material
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Vitaku E.Smith D. T.Njardarson J. T.Analysis of the Structural Diversity, Substitution Patterns, and Frequency of Nitrogen Heterocycles among U.S. FDA Approved Pharmaceuticals J. Med. Chem.20145724102571027410.1021/jm 501100 b 25255204 · doi ↗ · pubmed ↗
- 2Taylor R. D.Mac Coss M.Lawson A. D. G.Rings in Drugs J. Med. Chem.201457145845585910.1021/jm 401762524471928 · doi ↗ · pubmed ↗
- 3Shearer J.Castro J. L.Lawson A. D. G.Mac Coss M.Taylor R. D.Rings in Clinical Trials and Drugs: Present and Future J. Med. Chem.202265138699871210.1021/acs.jmedchem.2c 0047335730680 PMC 9289879 · doi ↗ · pubmed ↗
- 4Zakharychev V. V.Martsynkevich A. M.Development of novel pyridine-based agrochemicals: A review Advanced Agrochem.202541304810.1016/j.aac.2024.10.002 · doi ↗
- 5Marshall C. M.Federice J. G.Bell C. N.Cox P. B.Njardarson J. T.An Update on the Nitrogen Heterocycle Compositions and Properties of U.S. FDA-Approved Pharmaceuticals (2013–2023)J. Med. Chem.20246714116221165510.1021/acs.jmedchem.4c 0112238995264 · doi ↗ · pubmed ↗
- 6Zhang S.Ouyang Y.Gao Y.Li P.Design and Application of New Pyridine-Derived Chiral Ligands in Asymmetric Catalysis Acc. Chem. Res.202457695797010.1021/acs.accounts.3c 0080838446135 · doi ↗ · pubmed ↗
- 7Kwong H.-L.Yeung H.-L.Yeung C.-T.Lee W.-S.Lee C.-S.Wong W.-L.Chiral pyridine-containing ligands in asymmetric catalysis Coord. Chem. Rev.2007251172188222210.1016/j.ccr.2007.03.010 · doi ↗
- 8de Ruiter G.Lahav M.van der Boom M. E.Pyridine Coordination Chemistry for Molecular Assemblies on Surfaces Acc. Chem. Res.201447123407341610.1021/ar 500112 b 25350402 · doi ↗ · pubmed ↗