Molecular mechanism of resistance to lonafarnib conferred by mutations in the cysteine-rich region of respiratory syncytial virus fusion glycoprotein and discovery of a lonafarnib-derived antiviral PROTAC
Qi Yang, Bao Xue, Xianjie Qiu, Kaixin Yang, Jielin Tang, Anqi Zhou, Jingjing Zou, Yuhan Mao, Jiayi Zhong, Yuan Zhou, Wei Zhang, Qiong Zhang, Qingyu Xiao, Wei Tang, Zhiyu Li, Wencai Ye, Gang Zou, Wei Peng, Jinsai Shang, Xi Xu, Yixue Li, Xinwen Chen

TL;DR
This study identifies how respiratory syncytial virus becomes resistant to the antiviral lonafarnib and introduces a new drug strategy to prevent resistance.
Contribution
The paper discovers specific resistance mutations in RSV and designs a novel PROTAC compound to degrade the viral fusion protein.
Findings
Mutations at K394, K399, and T400 in RSV fusion protein confer high-level resistance to lonafarnib.
Resistance mutations reduce fusion protein stability and binding affinity for lonafarnib.
A lonafarnib-derived PROTAC compound effectively inhibits RSV replication by degrading the fusion protein.
Abstract
Lonafarnib, an oral antiviral that targets the fusion glycoprotein of respiratory syncytial virus (RSV), has demonstrated efficacy in vitro and in vivo. However, because the RSV has evolved to become resistant to other fusion inhibitors, there is a concern that the same could occur for lonafarnib. Here, we identified resistance to lonafarnib in the RSV A2 strain and a recent clinical isolate, RSV ON1, via in vitro selection at scale. Cell‒cell fusion and recombinant live RSV analysis confirmed that the mutations at K394, K399, and T400 of the cysteine-rich region of the fusion protein mediate high-level resistance. Lonafarnib resistance mutations also confer cross-resistance to other fusion inhibitors of clinical interest. All-atom molecular dynamics simulations revealed that these resistance mutations confer reduced stability to the fusion protein, thereby diminishing its binding…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
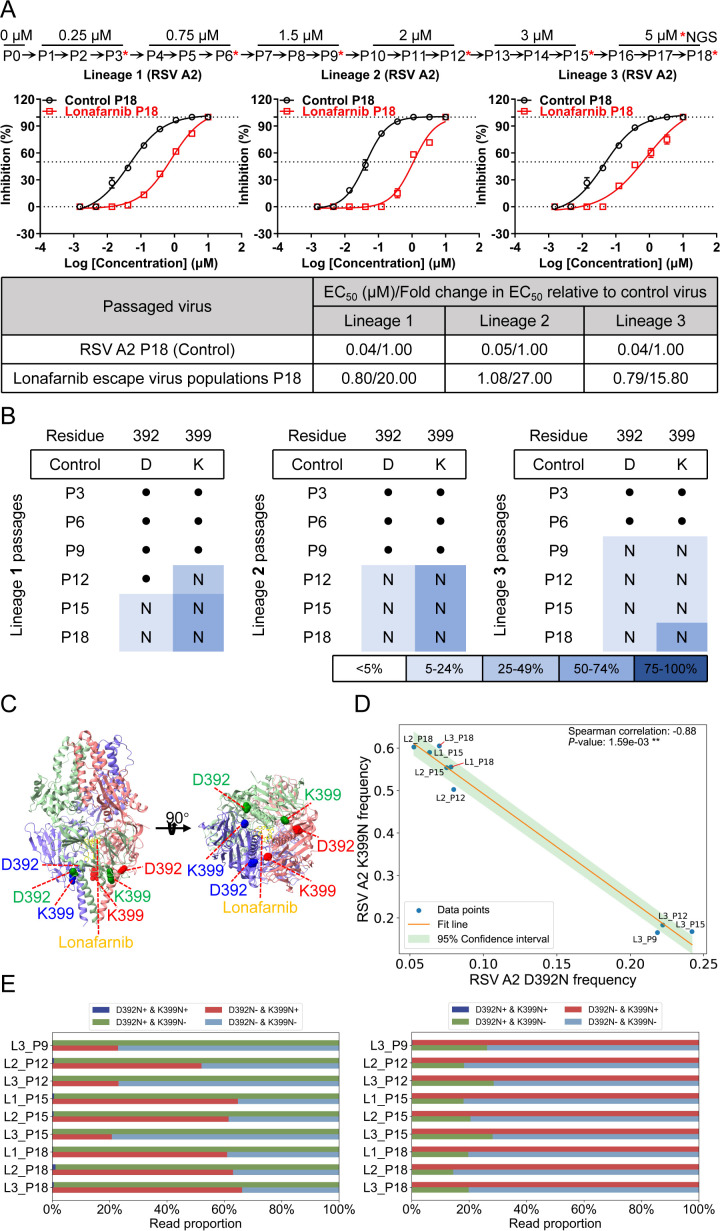 Fig 1
Fig 1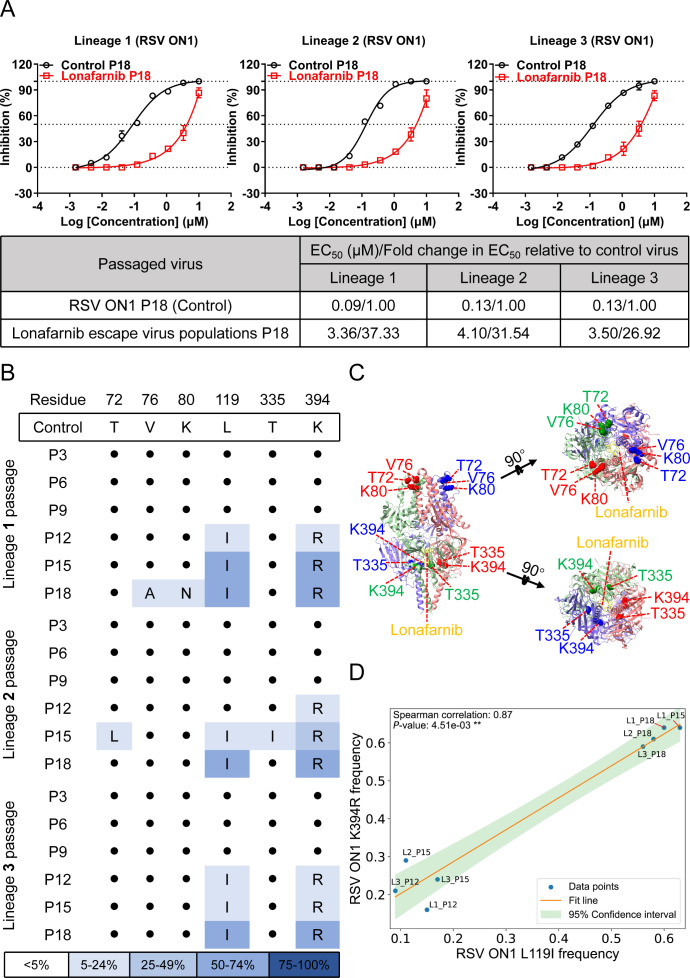 Fig 2
Fig 2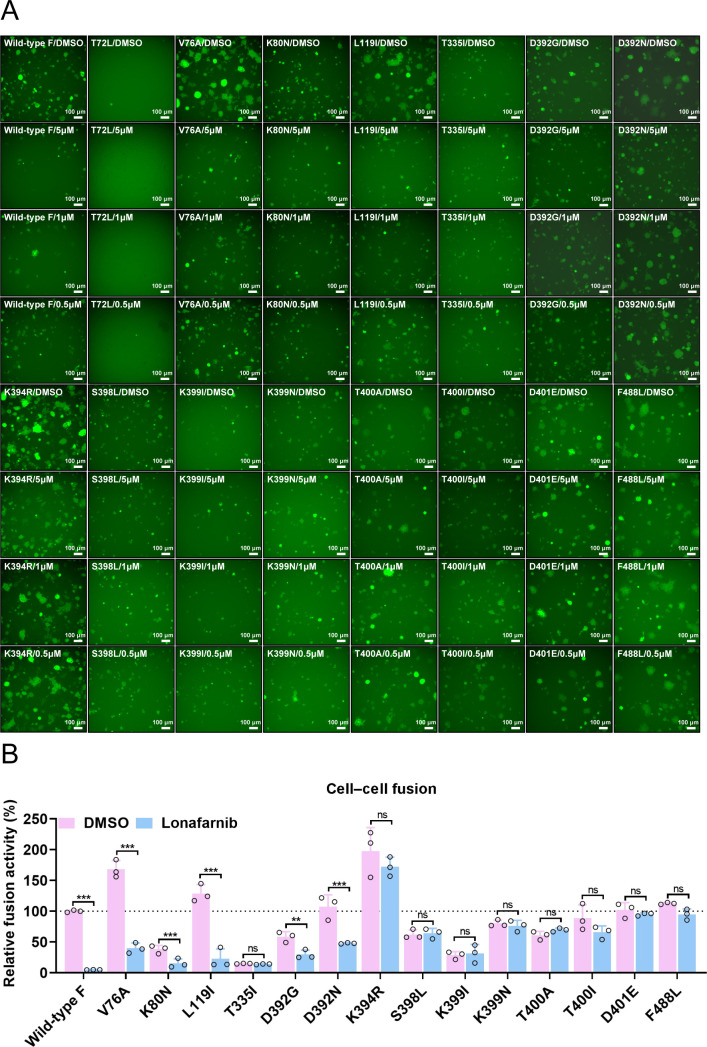 Fig 3
Fig 3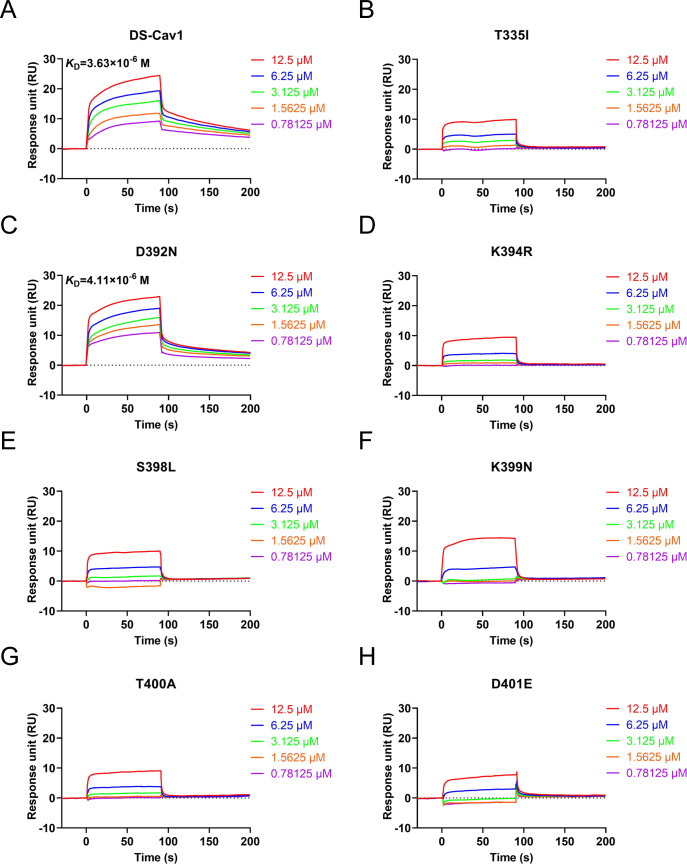 Fig 4
Fig 4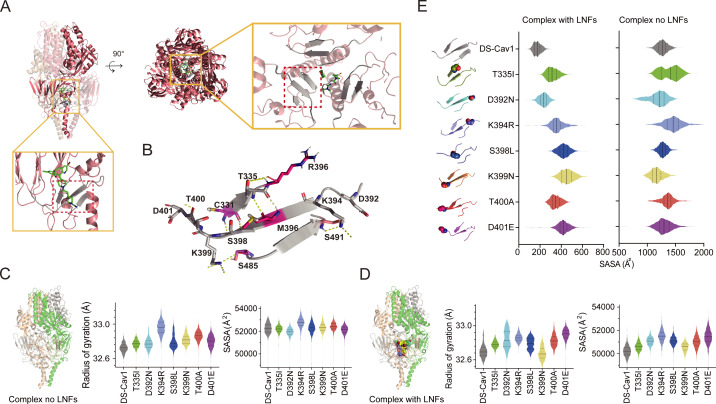 Fig 5
Fig 5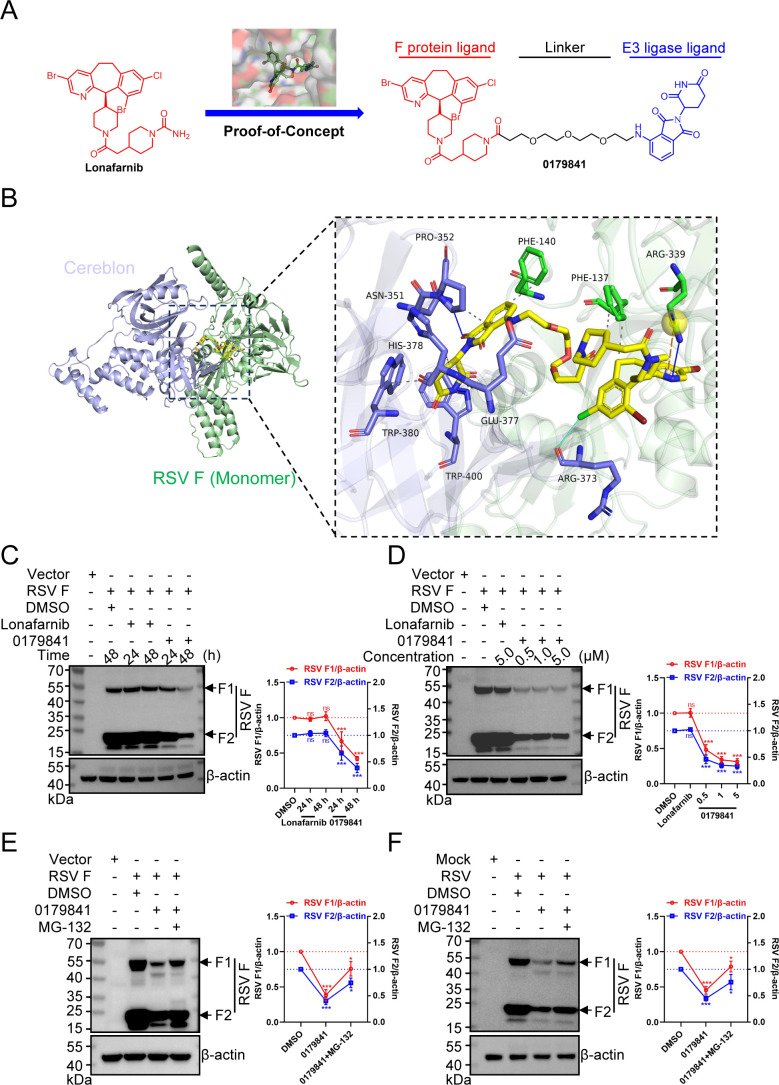 Fig 6
Fig 6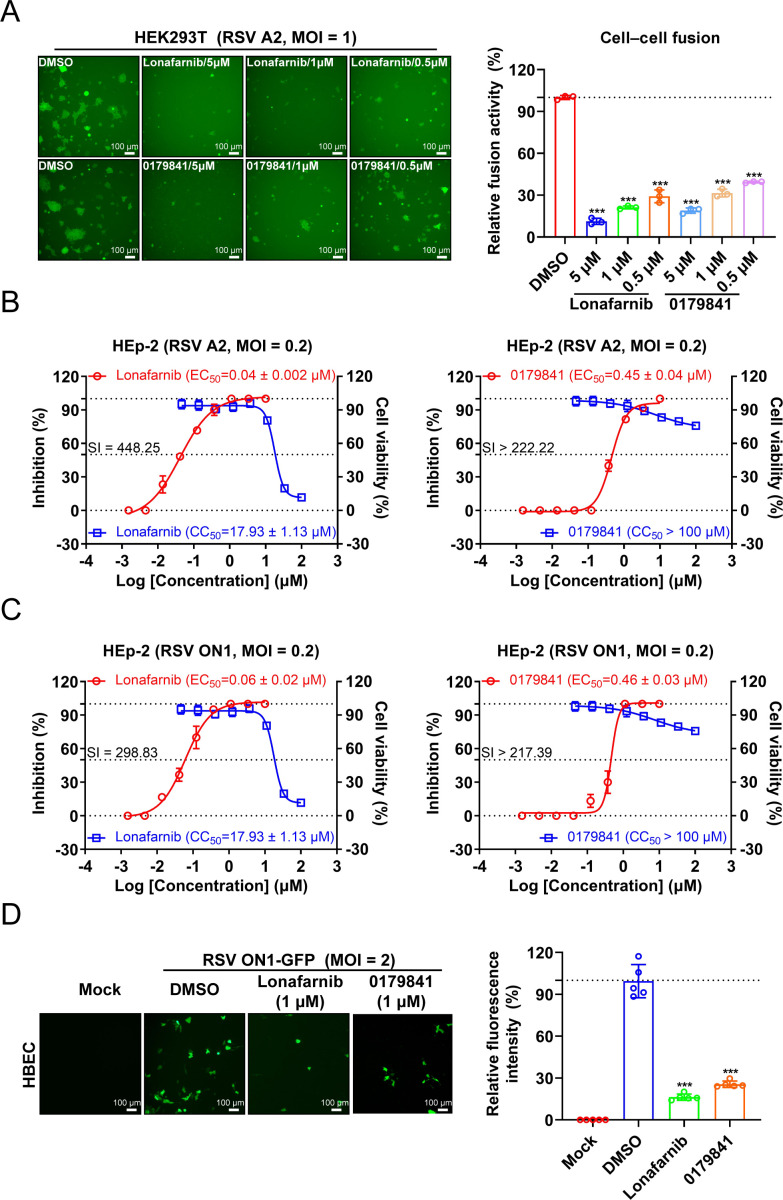 Fig 7
Fig 7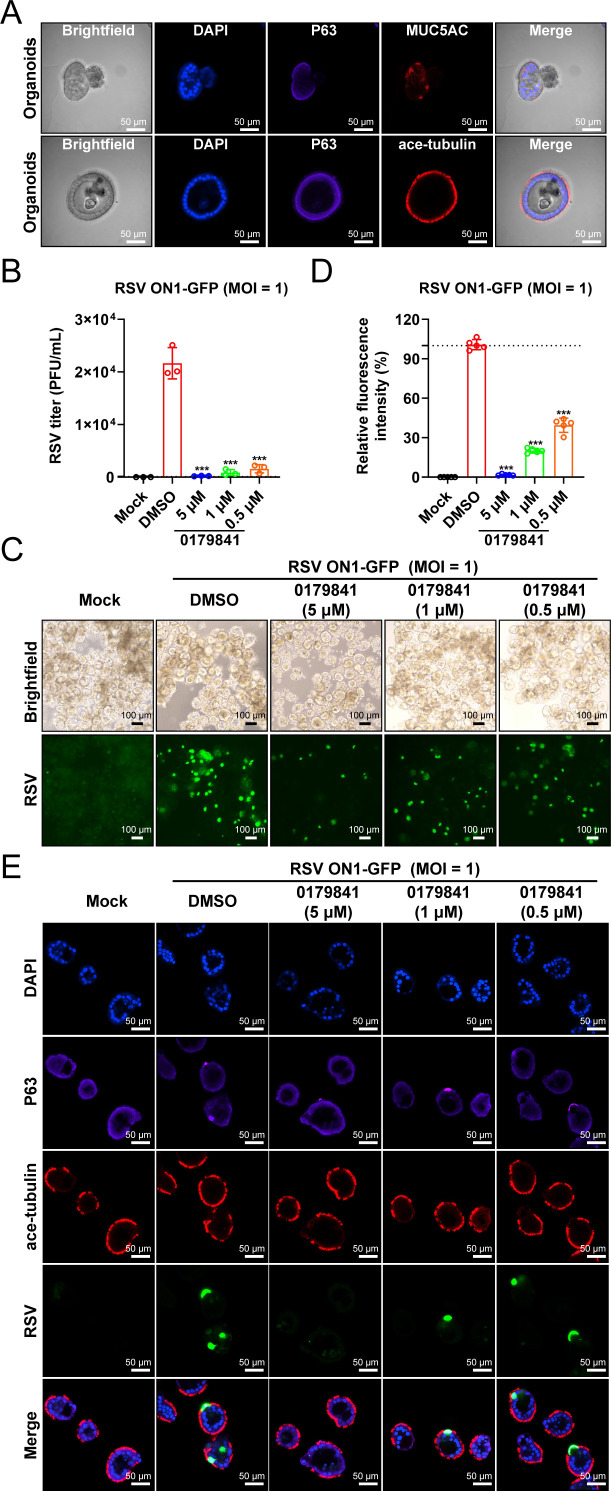 Fig 8
Fig 8| RSV strain | EC50 (μM) | Fold change in EC50 | EC90 (μM) | Fold change in EC90 |
|---|---|---|---|---|
| Wild-type | 0.046 ± 0.010 | 1.0 | 0.190 ± 0.120 | 1.0 |
| V127G | 0.044 ± 0.005 | 0.964 | 0.177 ± 0.038 | 0.935 |
| L141F | 0.079 ± 0.019 | 1.717 | 0.631 ± 0.125 | 3.326 |
| K394R | 5.127 ± 1.689 | 111.464 | 15.025 ± 7.331 | 79.202 |
| K399N | 0.450 ± 0.032 | 9.786 | 1.552 ± 0.277 | 8.183 |
| T400A | 1.522 ± 0.127 | 33.087 | 4.640 ± 0.440 | 24.458 |
| T400I | 1.506 ± 0.184 | 32.746 | 4.611 ± 0.663 | 24.308 |
| F488L | 0.700 ± 0.069 | 15.228 | 1.900 ± 0.160 | 9.992 |
| Inhibitor | EC50 (nM) | Fold resistance | EC50 (nM) | Fold resistance | EC50 (nM) | Fold resistance | |
|---|---|---|---|---|---|---|---|
| RSV | RSV K394R | RSV K399N | RSV T400A | ||||
| Lonafarnib | 45.6 | 3,839.0 | 84.3 | 405.6 | 8.9 | 1,117.0 | 24.5 |
| GS-5806 | 0.1 | 192.5 | 1,673.9 | 4.4 | 38.3 | 28.8 | 250.1 |
| AK0529 | 1.3 | 6,553.0 | 4,983.3 | 2,253.0 | 1,713.3 | 9,851.0 | 7,491.3 |
| RV521 | 0.1 | 178.9 | 2,462.8 | 0.4 | 5.4 | 12.6 | 173.6 |
| JNJ-53718678 | 0.7 | 10,130.0 | 15,582.2 | 248.5 | 382.2 | 11,650.0 | 17,920.3 |
- —Major Project of Guangzhou Laboratory
- —Major Project of Guangzhou Laboratory
- —Natural Science Foundation of Guangdong Provincehttp://dx.doi.org/10.13039/501100003453
- —National Natural Science Foundation of Chinahttp://dx.doi.org/10.13039/501100001809
- —National Natural Science Foundation of Chinahttp://dx.doi.org/10.13039/501100001809
- —Peal River Talent Recruitment Program
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsRespiratory viral infections research · Protein Degradation and Inhibitors · Virus-based gene therapy research
INTRODUCTION
Respiratory syncytial virus (RSV) is a common cause of acute lower respiratory tract infection in infants, older adults, and immunocompromised individuals (1, 2). Despite the clinical benefits of three approved RSV vaccines (Arexvy (3) [GSK]; Abrysvo (4) [Pfizer]; or mResvia (5) [Moderna]) and two monoclonal antibodies (palivizumab (6) [AstraZeneca] and nirsevimab (7) [AstraZeneca, Sanofi]), treatment remains limited to high-risk people, with approval in only a few countries and regions. Arexvy, Abrysvo, and mResvia are available for adults; however, no vaccines are approved for the prevention of RSV infection in infants or children (8). The Food and Drug Administration recently suspended all clinical trials of RSV vaccines in infants and young children following reports of severe illness in infants vaccinated with Moderna’s mRNA-based RSV vaccine (9). Palivizumab and nirsevimab are approved for the prevention but not the treatment of RSV disease (10).
There are currently no approved RSV-specific therapeutic small molecules available, but multiple clinical-stage small molecules, including fusion inhibitors targeting the viral fusion glycoprotein (F) (11–15) and replication inhibitors targeting the viral nucleoprotein (N) (16) or large polymerase subunit (L) (16–18), are currently available as drug candidates for treating RSV infections. These three viral proteins are essential for the viral life cycle and are well conserved among Orthopneumoviruses (19, 20). Nevertheless, the development of mutational escape and therapeutic resistance is still common for RSV (14, 15, 21–28), which is a particular concern in immunocompromised individuals.
The RSV F protein plays a critical role in mediating viral entry into host cells (20, 29) and induces the formation of syncytia between infected cells and neighboring uninfected cells (30, 31). Inhibition of viral entry and spread by blocking the exchange of a metastable state (prefusion) with a stable state (postfusion) of the RSV F protein during the fusion process has emerged as a promising treatment option for patients infected with RSV (19, 32). Currently, GS-5806 (14), AK0529 (15), RV521 (12), and JNJ-53718678 (13) are several structurally heterogeneous and potent fusion inhibitors that are being clinically developed for RSV infection treatment. However, all of these inhibitors have the potential to induce the emergence of resistant mutants both in vitro and in vivo (14, 15, 21, 22, 24, 26), as demonstrated by resistance-associated substitutions for GS-5806 (e.g., T400A, D486N) (21), JNJ-53718678 (e.g., L141W, D489Y) (22), AK0529 (e.g., D486N, D489A/V/Y) (24), and RV521 (e.g., D489Y) (26) in vitro, and the most frequent substitutions (e.g., F140I, L141F/W, S398L, K399N, T400A/I, D486N, or F488L) in the F protein to GS-5806 or AK0529 have already been identified in real-world cases (14, 15). Among the known mutations, D486N and D489Y have shown cross-resistance to various fusion inhibitors, and the emergence of both mutants is a potential concern for human health (33). Thus, new therapeutic options are still needed.
Lonafarnib, an orally active farnesyltransferase inhibitor, has demonstrated high therapeutic efficacy for Hutchinson‒Gilford progeria syndrome (HGPS) (34) and is a phase III candidate for hepatitis delta virus (HDV) therapy (35). Recently, drug repurposing screens conducted by our team and another research group identified lonafarnib as a potent RSV fusion inhibitor (36, 37). In a murine infection model, oral administration of lonafarnib resulted in a dose-dependent reduction in the RSV load and alleviation of lung damage. Thus, the repurposing of lonafarnib has significant potential in combating RSV infection and may offer a more accessible treatment option for patients with HGPS, HDV, or cancer comorbidities. However, whether RSV can evolve resistance mechanisms similar to those observed with other fusion inhibitors remains a concern.
The emergence of novel viruses and drug-resistant strains highlights the urgent need for innovative antiviral therapies (38). Targeted protein degradation (TPD) represents an innovative drug discovery strategy that eliminates proteins of interest (POIs) by harnessing the intracellular ubiquitin-proteasome system (UPS), macroautophagy, and endolysosomal pathways (39). Proteolysis-targeting chimera (PROTAC) molecules serve as classic representatives of TPD and typically comprise three key components: a ligand for the POI, a linker, and a ligand for the E3 ligase. By bridging the POI and the E3 ligase, PROTACs facilitate the ubiquitination of the POI, thereby triggering its degradation through the proteasome system (40). In recent years, substantial research efforts have been devoted to the development of antiviral drugs that target a variety of viruses via PROTAC technology (such as hepatitis B virus [41], hepatitis C virus [42], influenza virus [43, 44], and SARS-CoV-2 [45, 46]). However, the potential of using PROTAC modalities to target the F protein for degradation and thereby inhibit RSV infection remains largely unexplored.
Here, we identified the conserved cysteine-rich region of the RSV F protein as the critical site associated with resistance to lonafarnib in vitro. To investigate the underlying molecular mechanisms of RSV resistance to lonafarnib, we conducted surface plasmon resonance (SPR) and all-atom molecular dynamics (MD) simulation assays for each resistance mutation in the presence of lonafarnib. Furthermore, we applied PROTAC approaches to develop a lonafarnib-based virus-specific antiviral degrader that targets the viral F protein.
RESULTS
Screening for RSV resistance to lonafarnib
To identify resistance mutations against lonafarnib, the RSV A2 strain was passaged in HEp-2 cells under increasing drug concentrations, and viruses from every third passage were subjected to next-generation sequencing (NGS) (Fig. 1A and Methods). Triplicate lineages developed high-level resistance after 18 passages, exhibiting a mean 20.9-fold increase in half-maximal effective concentration (EC_50_) values compared with those of the control virus (Fig. 1A). NGS revealed that the D392N and K399N mutations appeared in the F gene of the RSV A2 strain in all three lineages, the K399N mutation of which occurred in a stepwise manner (Fig. 1B; Table S1). The D392N and K399N mutations are located in the 392–401 microdomain of the F protein and in immediate proximity to the lonafarnib-binding microdomain (486–489 residues) in prefusion F (Fig. 1C) (36, 37). Correlation analysis of the mutation frequencies with >5% mutation across all nine samples revealed a significant negative correlation (Spearman coefficient of −0.88, P < 0.01) (Fig. 1D). χ^2^ testing of paired-read distributions confirmed non-independence, indicating mutual exclusivity (Fig. 1E).
Identification of the resistance of the RSV A2 strain to lonafarnib. (A) Passaging scheme. HEp-2 cells were infected in triplicate with RSV A2 and passaged into fresh cells every 2 days for 18 passages, and the concentration of lonafarnib was increased every three passages. Changes in the EC50 during passaging of RSV A2 with lonafarnib. Validation of lonafarnib resistance in the indicated passages from each of the three lineages (1 [left], 2 [middle], and 3 [right]). The graphs shown are the mean values of three independent experiments. (B) Mutations in the F protein were found in the indicated passages from each lineage. The dots indicate the control virus at that residue. Mutations are shaded according to frequency (see Table S1 for exact frequencies). (C) Residues mutated with passaging are overlaid onto the DS-Cav1 structure with lonafarnib bound. The Cα of each mutated residue is denoted by a colored sphere. The DS-Cav1-lonafarnib complex was downloaded from PDB under accession code 8KG5. (D) Spearman correlation analysis between D392N and K399N mutations in the RSV A2 F protein. Correlation analysis of D392N and K399N mutations with frequencies > 5% between all nine samples. (E) Linkage relationship between D392N and K399N mutations. The reads of the D392N and K399N mutations were extracted from nine samples simultaneously with two mutations. The number of reads with only the D392N mutation and only the K399N mutation, as well as the number of reads with neither mutation and the simultaneous appearance of two mutations, were counted via a contingency table, and a χ2 test for independence on this contingency table was performed. The proportions of K399N mutations in the D392 control and D392N mutation reads (left), and the proportions of D392N mutations in the K399 control and K399N mutation reads (right) were calculated.
Parallel passaging of the clinical isolate RSV ON1 (18 passages, with triplicate lineages) in the presence of increasing concentrations of lonafanib yielded EC_50_ increases ranging from 26.9-fold to 37.3-fold (Fig. 2A). Six mutations in the RSV F gene were identified: T72L, V76A, K80N, L119I, T335I, and K394R (Fig. 2B; Table S2). Four mutations (T72L, V76A, K80N, and T335I) presented a low prevalence (<10% mean frequency): T72L (7.2% in lineage 2), V76A (8.0% in lineage 1), K80N (7.8% in lineage 1), and T335I (5.1% in lineage 2). These four mutations occurred in a non-stepwise manner. In contrast, L119I and K394R were conserved across lineages and detected in passages 15–18. The amino acid change at position 119 is in the 27-amino-acid peptide (p27), which is removed by furin during the post-translational processing of the F protein (47, 48). The 394th amino acid residue is located in the 392–401 microdomain of the F protein and is in immediate proximity to the lonafarnib-binding microdomain (486–489 residues) in prefusion F (Fig. 2C) (36, 37). Owing to the low mutation frequency and skip-like occurrence of these four sites (T72L, V76A, K80N, and T335I), we only conducted correlation analysis on L119I and K394R mutations with frequencies > 5% between all eight samples, which showed a significant positive correlation (Spearman coefficient of 0.87, P < 0.01) (Fig. 2D). Physical distance precluded read-level independence analysis between the L119I and K394R mutations.
Identification of the resistance of the RSV ON1 strain to lonafarnib. (A) HEp-2 cells were infected in triplicate with RSV ON1 and passaged into fresh cells every 2 days for 18 passages, and the concentration of lonafarnib was increased every three passages. Changes in the EC50 for the indicated passages from each of the three lineages were observed. Validation of lonafarnib resistance in the indicated passages from each of the three lineages (1 [left], 2 [middle], and 3 [right]) was performed. The graphs shown are the mean values of three independent experiments. (B) Mutations in the F protein were found in the indicated passages from each lineage. The dots indicate the control virus at that residue. Mutations are shaded according to frequency (see Table S2 for exact frequencies). (C) Residues mutated with passaging are overlaid onto the DS-Cav1 structure with lonafarnib bound. The Cα of each mutated residue is denoted by a colored sphere. The DS-Cav1-lonafarnib complex was downloaded from PDB under accession code 8KG5. (D) Spearman correlation analysis between L119I and K394R mutations of the RSV ON1 F protein. Correlation analysis of L119I and K394R mutations with frequencies > 5% across all 8 samples.
Overall, selection at the scale revealed that RSV can lead to the development of lonafarnib resistance. The differences in mutation sites could arise in the RSV A2 and ON1 strains, and all these mutations were not situated in the lonafarnib-binding sites; rather, several mutations in the 392–401 microdomain of the RSV F protein were preferred.
Characterization of lonafarnib-resistant mutants
To comprehensively investigate which mutations are responsible for lonafarnib resistance, we constructed eight mutations identified in this study (T72L, V76A, K80N, L119I, T335I, D392N, K394R, and K399N) and other fusion inhibitors reported resistance-associated substitutions (D392G/N, K394R, S398L, K399I/N, T400A/I, and D401E) in the 392–401 microdomain (15, 21, 36, 49–52). Using a GFP-split fusion model for cell‒cell fusion mediated by the RSV F protein (Fig. S1), we observed that the substitutions in RSV F resulted in its loss (T72L), decrease (K80N, T335I, and K399I), or increase (V76A, L119I, and K394R) in membrane fusion ability (Fig. 3A and B). Other substitutions (D392G/N, S398L, K399N, T400A/I, D401E, and F488L) of RSV F have the same ability to induce fusion as the wild-type F protein does. F488 serves as a binding site for lonafarnib (37) and acts as an experimental positive control. F488L alteration decreased drug binding and resulted in a loss of inhibitory activity. Compared with wild-type F, lonafarnib significantly inhibited V76A, K80N, L119I, and D392G/N mutant-induced cell‒cell fusion activity, suggesting that alterations at these sites are largely not responsible for lonafarnib resistance (Fig. 3A and B). Lonafarnib did not or only slightly inhibit the fusion activity induced by T335I, S398L, K394R, K399I/N, T400A/I, and D401E, indicating that these mutations in the cysteine-rich region might lead to the resistance of RSV to lonafarnib.
*Validation of the resistance of the identified mutations to lonafarnib via a cell‒cell fusion assay. (A) Effects of lonafarnib on wild-type F- or F mutation-mediated cell‒cell fusion. All mutations identified in this study (T72L, V76A, K80N, L119I, T335I, D392N, K394R, and K399N) and other resistance-associated substitutions (D392G/N, K394R, S398L, K399I/N, T400A/I, and D401E) reported for fusion inhibitors in the 392–401 microdomain were constructed. F488 serves as a binding site for lonafarnib and acts as an experimental positive control. HEK293T GFP-split cells were transfected with plasmids expressing wild-type F or F mutants and treated with 0.1% DMSO or lonafarnib (5, 1, and 0.5 µM). The cell‒cell fusion activity was determined by GFP expression 24 h post-transfection. Images represent three biological replicates comprising nine fields each. (B) Quantification of cell‒cell fusion activity was performed via ImageJ analysis software. For statistical analysis, nonparametric Mann‒Whitney tests were used for comparisons. The results are representative of three random experiments. The error bars indicate the means ± SDs of three technical replicates. **P < 0.01, **P < 0.001; ns, not significant.
Next, we investigated mutations in recombinant live RSV carrying single F protein mutations. Seven mutant viruses (V127G, L141F, K394R, K399N, T400A, T400I, and F488L) were used in this study, with V127G and L141F mutant viruses used as the experimental negative controls and F488L as the positive control. The infectious titer produced by HEp-2 cells infected with wild-type RSV increased faster than did the titer produced by the viruses with mutations (Fig. S2A), and after 90 h, the titer of the wild-type virus was almost 10-fold greater than the titer achieved by viruses with mutations except for V127G (Fig. S2A). Among the individual mutants tested against lonafarnib, the infection of the wild-type and V127G mutant viruses was effectively inhibited; L141F showed only minimal resistance (~1.7-fold to 3.3-fold); K399N (~9.8-fold increase in the EC_50_ and ~8.2-fold increase in the EC_90_), and F488L (~15.2-fold increase in the EC_50_ and ~10-fold increase in the EC_90_) presented high-level resistance relative to that of the wild-type virus; the increased resistance caused by T400A (~33-fold increase in the EC_50_ and ~24-fold increase in the EC_90_) was approximately equivalent to that caused by T400I; K394R (~111.5-fold increase in the EC_50_ and ~79.2-fold increase in the EC_90_) was the most resistant (Table 1; Fig. S2B). Together, these results indicate that the K394R, K399N, and T400A/I mutations in the cysteine-rich region of the F protein favor RSV resistance to lonafarnib.
Furthermore, we investigated mutant viruses (K394R, K399N, and T400A) against other clinically relevant fusion inhibitors (e.g., GS-5806 (14), AK0529 (15), RV521 (12), and JNJ-53718678 (13)) for cross-resistance. All three of these mutant viruses exhibited considerable resistance to the above four fusion inhibitors, but K399N resulted in only slight resistance to RV521 (Table 2; Fig. S3). Selection for lonafarnib resistance can clearly yield mutations that confer cross-resistance to other inhibitors of clinical interest as well.
Mechanism of lonafarnib resistance
To investigate the mechanisms underlying RSV resistance to lonafarnib in the cysteine-rich region of the F protein, we expressed prefusion-stabilized RSV F variant DS-Cav1 (19) and its mutants (T335I, D392N, K394R, S398L, K399N, T400A, and D401E) in HEK293F cells (Fig. S4A and B). SPR analysis revealed that the mutants (T335I, K394R, S398L, K399N, T400A, and D401E) significantly decreased the binding affinity for lonafarnib (Fig. 4B and D through H). Compared with DS-Cav1, the D392N mutant retained the ability to bind to lonafarnib but presented slightly lower affinities (Fig. 4A and C). Next, all-atom MD simulations were conducted on DS-Cav1 and these mutants. The simulations were executed for 500 ns and replicated three times (Fig. S5) under two different conditions, where the prefusion F trimer complex was present in the absence or presence of lonafarnib. These residues are located around the central cavity, which includes a cysteine-rich region and heptad repeat B (Fig. 5A and B). The DS-Cav1 variant presented three stable β-sheet conformations in the main cluster structure (Fig. 5A and B), and these β-sheets formed at residues 331–335, 394–398, and 486–488 in the DS-Cav1 cluster structure. As shown, these residues engage in polar contacts: C331 with S398, T335 with M396, K394 with S491, and K399 with S485 (Fig. 5B). The formamide group of the lonafarnib chemical structure is crucial for binding by providing polar interactions, which form hydrogen bonds with residues T397, S398, D486, and E487 (37). The radius of gyration (Rg) directly measures protein stability, showing its folding state and compactness. The solvent-accessible surface area (SASA) reflects stability by indicating the protection of the hydrophobic core. Both parameters enable a comprehensive assessment of protein stability. MD simulations revealed that the DS-Cav1 and D392N mutations were the most stable among all the simulation cases (Fig. 5C). Conversely, the K394R mutation resulted in the highest values of both Rg and SASA, indicating a decrease in stability compared with those of DS-Cav1 and other mutants. Notably, even in the presence of lonafarnib, DS-Cav1 still maintained its status as the most stable (Fig. 5D). Similarly, K394R also presented the highest Rg and SASA values, indicating its relatively lower stability than the other mutations.
Binding affinity of prefusion-stabilized RSV F variant DS-Cav1 and its mutants for lonafarnib. (A–H) BIAcore diagrams of DS-Cav1, T335I, D392N, K394R, S398L, K399N, T400A, and D401E binding to lonafarnib. These mutations D392G/N, K399I/N, and T400A/I exhibit no significant difference in their anti-fusion capabilities in cell‒cell fusion assays; thus, only one mutation at the same site was selected for SPR assays. The graphs shown are representative of three independent experiments.
All-atom dynamic simulations of DS-Cav1 and seven mutant residues (T335I, D392N, K394R, S398L, K399N, T400A, and D401E) of the RSV F protein. (A) Schematic view of the main cluster of the molecular dynamics simulation. The yellow rectangular box outlines the central cavity region, and the red dotted-line box outlines the regions of the three stable β-sheet residues (B). Complex MD models without (C) or with (D) lonafarnibs (LNFs). The radius of gyration (Rg) directly measures protein stability, showing its folding state and compactness. The solvent-accessible surface area (SASA) reflects stability by indicating the protection of the hydrophobic core. Both parameters enable a comprehensive assessment of protein stability. (E) Statistical analysis of the SASA values of the three stable beta-sheet residues with DS-Cav1 and seven different cases of mutant residues in the RSV F complex with or without LNFs.
To investigate the effects of lonafarnib on various mutations, we analyzed the SASA values of residues surrounding the central cavity, which are residues 333–335, 392–401, and 486–488. The results demonstrated that the DS-Cav1 and D392N mutants presented increased binding affinity for lonafarnib (Fig. 5E). In contrast, other mutations significantly weakened the interaction between the F protein and lonafarnib. Notably, in the RSV F complex model with the K399N mutation, relatively low Rg and SASA values were observed (Fig. 5D), suggesting that lonafarnib may still bind to this mutation. However, the central cavity area of the K399N mutation appears less stable than that of the DS-Cav1 and D392N mutations (Fig. 5E). These findings are consistent with previous experimental data (Fig. 4), validating the role of mutations in modulating the lonafarnib interaction within the molecular mechanisms.
Compound 0179841-induced degradation of the F protein confers an anti-RSV effect
The resistance mutations in the central cavity region of the RSV F protein confer resistance by structurally destabilizing the protein and reducing the binding affinity of lonafarnib. Crucially, the long-range impact of these mutations on the β-sheet network and hydrophobic cavity conformation underscores how current fusion inhibitors remain susceptible to allosteric resistance mechanisms. As most small-molecule candidates targeting the RSV F protein pose a considerable risk of inducing drug resistance mutations, these findings highlight the pressing need for new approaches to address this vulnerability proactively and overcome the barrier to resistance development. Here, we applied the PROTAC approach to develop a lonafarnib-based antiviral degrader, 0179841, that targets the RSV F protein. The molecule 0179841 anchors to the hydrophobic domain of the RSV F protein through its lonafarnib module and recruits the E3 ubiquitin ligase complex via the cereblon ligand, thereby enabling ubiquitination-dependent degradation of the target (Fig. 6A; see synthetic details in Fig. S6 and S7). Computational docking predicted the binding conformation between PROTAC 0179841 and both the RSV F protein and the cereblon protein. The best binding poses were identified on the basis of geometric and energy matching, yielding a binding score of −11.38. The results showed that compound 0179841 interacts directly with three residues (Phe137, Phe140, and Arg339) in the monomeric F protein and two residues (Asn351 and Trp380) in the cereblon protein (Fig. 6B). Four sets of experiments were conducted to assess the selective degradation and inhibitory effects of compound 0179841 on the RSV F protein. In HEK293T cells expressing the RSV F protein, we measured the protein levels of F with and without compound 0179841 treatment. We found that compound 0179841-induced degradation of the F protein is time- and concentration-dependent (Fig. 6C and D), which appears to occur through proteasomal degradation pathways (Fig. 6E), but lonafarnib did not affect F protein levels (Fig. 6C and D). Notably, compound 0179841 also induced significant degradation of the F protein during RSV infection (Fig. 6F).
*The PROTAC 0179841 depletes the RSV F protein. (A) Structure of virus-specific and host-directed antivirals utilizing targeted protein degradation, comprising a lonafarnib-derived F protein ligand (red), a PEG-based flexible linker (black), and a cereblon-targeting E3 ubiquitin ligase ligand (blue). (B) Docking model of the binding of compound 0179841 to the RSV F monomer and cereblon. The RSV F monomer is depicted as green sticks, the cereblon is depicted as blue sticks, and compound 0179841 is depicted as yellow sticks. (C–E) HEK293T cells were transfected with F plasmids, and the cell lysates were analyzed via western blotting with antibodies against RSV F or β-actin (left) and densitometric analysis (right). (C) The transfected cells were treated with 0.1% DMSO, 1 µM lonafarnib, or compound 0179841 for the indicated periods post-transfection. (D) The transfected cells were treated with 0.1% DMSO or the indicated compounds at the indicated concentrations for 48 h. (E) The transfected cells were treated with 0.1% DMSO or 1 µM compound 0179841 for 48 h, and the cells were then treated with either 0.1% DMSO or 20 µM MG-132 for 8 h before sample collection. (F) HEp-2 cells were infected with RSV (MOI = 10) and subsequently treated with either 0.1% DMSO or 1 µM compound 0179841 for 18 h. Prior to sample collection, the cells were further treated with either 0.1% DMSO or 20 µM MG-132 for 8 h. The cell lysates were then analyzed by western blotting using antibodies against RSV F and β-actin (left) and densitometric analysis (right). The graphs shown are representative of three independent experiments, and the error bars indicate the means ± SDs derived from these three independent experiments. *P < 0.05, **P < 0.001; ns, not significant (one-way ANOVA with Dunnett’s post hoc test).
Next, using the GFP-split fusion model for cell‒cell fusion mediated by RSV, we observed that the compound 0179841 significantly inhibited RSV-induced cell‒cell fusion activity (Fig. 7A). Compound 0179841 also effectively inhibited the replication of the RSV A2 and ON1 strains in HEp-2 cells without affecting cell viability, with EC_50_ values of 0.45 ± 0.04 µM and 0.46 ± 0.03 µM, respectively (Fig. 7B and C). The selectivity index (SI) of compound 0179841 exceeds 217.39 in HEp-2 cells, indicating its safety at the cellular level. Furthermore, we evaluated the in vitro antiviral efficacy of compound 0179841 against RSV ON1-GFP in primary human bronchial epithelial cells (HBECs). Treatment with compound 0179841 resulted in a significant decrease in the RSV infection level, as evidenced by marked reductions in the GFP intensity (Fig. 7D). Taken together, these results suggest that compound 0179841 can target the F protein for degradation and confer antiviral activity.
*In vitro anti-RSV activity of compound 0179841. (A) Effect of compound 0179841 on RSV-mediated cell‒cell fusion. HEK293T GFP-split cells were infected with RSV A2 (MOI = 1) and treated with lonafarnib or compound 0179841 at the indicated concentrations (5, 1, and 0.5 µM) or with 0.1% DMSO as a control. Representative images of cell‒cell fusion at 24 h post-infection are shown (left). The quantification of cell‒cell fusion activity according to the GFP intensity in three random fields per experiment was performed via ImageJ analysis software, and the data were normalized to the intensity in the DMSO condition (right). (B, C) HEp-2 cells were infected with the RSV A2 (B) or ON1 (C) strain (MOI = 0.2) and treated with the indicated concentrations of the indicated compounds for 72 h post-infection. The inhibitory effects of the indicated compounds on RSV were quantified via FFA. The EC50 represents the concentration required to inhibit RSV infection by 50%. The cytotoxicity of the indicated compounds to HEp-2 cells was measured via cell viability assays. The selectivity index (SI) = CC50/EC50 was calculated. (D) Effects of compound 0179841 on RSV infection in primary human bronchial epithelial cells (HBECs). HBECs were infected at an MOI of 2 with the RSV ON1-GFP strain and treated with either 1 µM lonafarnib or compound 0179841. The infected cells were subsequently visualized at 72 h post-infection (left). The GFP intensity in five randomly selected fields per experiment was measured via ImageJ analysis software and normalized to the intensity in the DMSO condition (right). The graphs shown are representative of three independent experiments. The error bars represent the means ± SDs from three technical replicates in panel (A) and five technical replicates in panel (D). The differences between the experimental and DMSO groups were determined by Student’s t-test for comparing two groups of data (**P < 0.001).
Compound 0179841 inhibits RSV infection in human respiratory organoids
Human respiratory organoids are widely utilized as model systems for investigating human respiratory viral pathogenesis and supporting pharmaceutical research (53–57). Here, we differentiated HBECs into mature, physiologically representative apical-out airway organoids. These organoids contain basal, secretory (goblet), and outward-facing ciliated cells, mimicking the apical surface of the airway epithelium in vivo (Fig. 8A). Next, we evaluated the efficacy of compound 0179841 against RSV in the human airway organoid model. Compound 0179841 significantly reduced viral loads and protein expression in a dose-dependent manner in lung airway organoids (Fig. 8B through E). Compared with the DMSO control, compound 0179841 at a concentration of 5 µM reduced viral titers by more than 100-fold (Fig. 8B). Additionally, the GFP intensity demonstrated a pronounced antiviral effect of compound 0179841 against RSV ON1-GFP (Fig. 8C through E). These data demonstrate that compound 0179841 can inhibit RSV infection in a physiologically relevant organoid system.
*Evaluation of the antiviral activity of compound 0179841 in a human airway organoid model. (A) Establishment of HBEC-derived apical-out airway organoids. Representative white-light images of organoids cultured over the long term are presented, along with confocal images demonstrating the expression of respiratory epithelial markers. The confocal images illustrate the presence and spatial distribution of distinct cell types within the organoids, as identified by their specific markers: basal cells labeled with P63 (purple), goblet cells labeled with MUC5AC (green), and ciliated cells labeled with acetylated tubulin (red). (B–E) Organoids were infected with RSV ON1-GFP at an MOI of 1 and cultured in media supplemented with 0.1% DMSO or compound 0179841 at the indicated concentrations (5, 1, and 0.5 µM) for 96 h. (B) The cell culture supernatant of infected organoids was collected, and a TCID50 assay was performed. (C) The RSV ON1-GFP-infected organoids were visualized via microscopy. (D) The GFP intensity in five randomly selected fields per treatment was measured using ImageJ analysis software and normalized to the intensity in the DMSO condition. (E) The RSV ON1-GFP-infected organoids were visualized using a confocal microscope. The graphs shown are representative of three independent experiments. The differences between the experimental and DMSO groups were determined by Student’s t-test for comparing two groups of data (**P < 0.001).
DISCUSSION
Fusion inhibitor-based interventions for RSV are facing increasing resistance development in vitro and in vivo, underscoring the need for new antivirals or alternative modes of action. Lonafarnib, a novel antiviral that targets the F protein, has shown efficacy in a BALB/c mouse infection model (36, 37). Given the adaptations that RSV has already exhibited to other fusion inhibitors, understanding potential lonafarnib resistance mechanisms is crucial. The results presented herein demonstrate that multiple different mutational residues in the fusion protein of diverse RSV strains are associated with lonafarnib resistance, with a notable preference for mutations in the cysteine-rich region. Mechanistic studies indicate that resistance mutations occupy internal sites within the prefusion RSV F protein, likely destabilizing it and reducing its binding affinity for lonafarnib. Importantly, we designed and identified a lonafarnib-based antiviral degrader, compound 0179841, which inhibits RSV infection by inducing F protein degradation.
The T335I and T400A mutations in the F protein of the RSV Long strain (rHRSV-A-GFP), which were previously linked to lonafarnib resistance, were confirmed (36). Here, large-scale passaging of RSV with lonafarnib selected for resistance mutations identified K399N in the RSV A2 strain and K394R in the clinically dominant RSV ON1 strain as prevalent substitutions (>5% mutation frequency, emerging in a stepwise manner). The differing mutations observed among these strains emphasize the complexity and variety of pathways leading to lonafarnib resistance, although it is not yet clear why certain mutations are specific to a particular strain. Although the mutation sites may vary among RSV strains, all the resulting amino acid substitutions are confined to the relatively conserved cysteine-rich region of the F protein. Cell‒cell fusion analysis of these mutants and other reported fusion inhibitors confirmed resistance-associated substitutions (D392G (49), S398L (50, 58), K399I (51), T400I (52), and D401E (33)) in the region in which these amino acid mutations in the cysteine-rich region mediate high-level resistance to lonafarnib, with the exception of D392G/N (Fig. 3), which was further validated via several recombinant live RSV strains carrying single F gene mutations (Table 1). Interestingly, the D392N and K399N mutations in the RSV A2 F protein were significantly negatively correlated, suggesting that they are not independent (Fig. 1E). These findings seem to indicate that lonafarnib could induce the D392N mutation to counteract mutations at other sites in the cysteine-rich region.
Furthermore, lonafarnib resistance mutations (K394R, K399N, and T400A) confer cross-resistance to other clinically relevant fusion inhibitors (e.g., GS-5806 (14), AK0529 (15), RV521 (12), and JNJ-53718678 (13)) (Table 2). GS-5806 (14), AK0529 (24), RV521 (26), and JNJ-53718678 (59) bind to a hydrophobic pocket in the F protein that is rich in phenylalanine residues (Phe488, Phe137, and Phe140). Our recently resolved cryo-EM structure of the F protein–lonafarnib complex revealed that lonafarnib occupies the same hydrophobic cavity and β-sheet network as these inhibitors do and additionally forms hydrogen bonds with phenylalanine residues (37). This binding pocket is proximal to residues K394, K399, and T400. Collectively, these structural insights explain why the K394R, K399N, and T400A mutations confer cross-resistance to multiple fusion inhibitors, including lonafarnib. Consequently, developing effective RSV F protein inhibitors or new therapeutic options against these mutants is urgently needed before the global spread of mutant strains.
Resistance mutations to RSV fusion inhibitors are predominantly located in a central cavity adjacent to the fusion peptide within the prefusion F trimer (32, 33). These regions include (i) the fusion peptide (amino acid residues 140–144), (ii) the cysteine-rich region (residues 392–401), and (iii) the heptad repeat B (residues 486–489). Resistance can arise through direct or indirect mechanisms. Direct mechanisms include mutations at the residues that interact directly with the inhibitors (e.g., D486N and F488L) or hindrance of the conformational changes required for inhibitor binding (e.g., D489Y) (32). Indirect mechanisms alter F protein stability and triggering rates (e.g., K394R, T400A, D401E, and D489E), narrowing the window for effective inhibitor binding (33). Mutations associated with lonafarnib-resistant RSV infection are located mainly in the cysteine-rich region of the F protein (Fig. 1 to 3; Table 1). These resistance-associated substitutions result in a low binding affinity by interfering with the binding of the drug to the hydrophobic pocket (Fig. 4) but do not participate directly in the binding of lonafarnib to the F protein (37). Importantly, the SPR experiments utilized a stabilized prefusion F protein; consequently, mutations that exert allosteric effects may not be detectable in this system. All-atom MD simulations revealed that mutations affect protein stability (Fig. 5). The lower Rg and SASA values in DS-Cav1 and D392N indicate greater stability. In contrast, other mutations, such as K394R, presented the highest SASA and Rg values, indicating decreased stability. This destabilization appears to disrupt the hydrophobic cavity, and the β-sheet network is critical for inhibitor binding, even for compounds that target nonoverlapping sites. Notably, mutations such as K394R in the cysteine-rich region (aa 392–401) induce conformational changes through interconnected structural motifs (e.g., β-sheets at residues 331–335 and 486–488), thereby indirectly altering the fusion peptide and HRB regions. It is worth noting that the K394R mutant confers resistance to lonafarnib by destabilizing the F protein and increasing its membrane fusion activity ([Fig. 3 and 5](#F3 F5)), along with cross-resistance to other fusion inhibitors (Table 2) (60). Therefore, it is conceivable that such an RSV mutant carrying the K394R mutation in the F protein may pose a potential future threat to lonafarnib-based therapeutics. This concern necessitates the development of next-generation inhibitors, or their combination with other agents, to treat RSV infection more effectively and prevent the emergence of drug-resistant RSV.
Since the emergence of PROTAC technology in 2001 (61), it has revolutionized drug discovery by enabling the degradation of disease-causing proteins. Over the past two decades, cancer research has undergone significant breakthroughs through the use of PROTACs to target “undruggable” oncoproteins (62). More recently, this approach has been extended to antiviral research, in which unique protein degradation pathways have been utilized to combat viral infections (46, 63). Conventional small-molecule inhibitors targeting the RSV F protein carry inherent risks of developing resistance, as demonstrated by the rapid emergence of escape mutations under selective pressure (14, 15, 21–26). To preemptively address this limitation, we pursued a degradation-based strategy using PROTACs. This approach aims to eliminate the target protein rather than modulate its activity, thereby reducing the opportunities for resistance mutations to confer survival advantages. In this study, we explored PROTAC modalities that target the RSV F protein for degradation to achieve virus-specific antiviral effects. The identification of the small-molecule degrader 0179841 demonstrates the potential for developing and rationally designing antivirals via the PROTAC platform. Compound 0179841 is a heterobifunctional small molecule composed of two ligands connected by a linker: lonafarnib (which recruits the F protein) and cereblon (which recruits E3 ubiquitin ligase) (Fig. 6). Simultaneous binding of both the F protein and the E3 ligase by compound 0179841 induces ubiquitylation of the F protein, leading to its subsequent degradation via the ubiquitin-proteasome system. Although compound 0179841 exhibited modest antiviral effects against RSV replication in vitro (Fig. 7 and 8), this proof-of-concept research is encouraging. Future studies will focus on optimizing PROTAC degrader sequences through structure‒activity relationship (SAR) analysis to enhance anti-RSV efficacy, pharmacokinetic properties, and immunogenicity profiles. These attributes are dictated by sequence architecture, length, and backbone configuration, which critically influence ternary complex formation and target engagement. Additionally, given that lonafarnib binds to the monomeric F protein and trimeric prefusion F protein (37), compound 0179841 may also bind inside the central cavity of the prefusion F protein to block its conformational change in addition to the degradation of the monomeric F protein. Although the dependence of compound 0179841 on lonafarnib binding means that it cannot directly overcome lonafarnib-associated resistance, as resistant mutations reduce the lonafarnib-F protein interaction (Fig. 4), it operates via a distinct antiviral mechanism—targeted degradation. Therefore, the resistance profile of 0179841 against RSV may plausibly differ from that of lonafarnib. Such insights would be invaluable for the development of novel antiviral therapeutics targeting this specific interaction, potentially offering a viable strategy against RSV.
Limitations of the study
Sequencing of escape populations revealed L119I and K394R mutations in the F gene of RSV ON1 in response to lonafarnib, and a correlation analysis of the two mutation frequencies revealed a significant positive correlation. However, the L119 residue is located in the p27 peptide, which is removed by furin during the post-translational processing of the F protein (47, 48); thus, we were unable to verify its role in drug resistance at the protein level. Additionally, our data suggest that at the doses and within the short time frames we utilized, the replication of RSV is sensitive to compound 0179841-mediated F protein degradation in vitro. However, in vivo validation in mice is limited for the following reasons: CRBN exhibits notable species-specific differences between mice and humans, especially in terms of substrate recognition and binding properties. A humanized CRBN (hCRBN) mouse model is therefore needed. Moreover, as a virus-directed strategy targeting proteostasis, further improvement of the EC_50_ against RSV in vitro, as well as analyses of the long-term pharmacokinetics, toxicity profiles, and delivery efficiency to lung tissue of compound 0179841 in an in vivo context, is still necessary.
MATERIALS AND METHODS
Cells, viruses, and compounds
HEp-2 cells (cat#CCL-23, RRID: CVCL_1906) and HEK293T cells (cat#CRL-3216, RRID: CVCL_0063) were purchased from the American Type Culture Collection (ATCC) and cultured in DMEM medium (HyClone, cat#SH30022.01) supplemented with 10% FBS (Thermo Fisher Scientific, cat#10099141), 2% HEPES (Thermo Fisher Scientific, cat#15630080), and 1% penicillin-streptomycin (Life Technologies, cat#15140122) at 37°C and 5% CO_2_ in a humidified incubator. HEK293F cell lines (RRID: CVCL_6642) were provided by Professor Xiaoli Xiong from Guangzhou Institutes of Biomedicine and Health and were propagated in serum-free SMM 293-TII Expression Medium (Sino Biological Inc., cat#M293TII) using orbital shakers (160 rpm) under equivalent temperature and CO₂ conditions. Human bronchial epithelial cells (HBECs, cat#CP-H009) were isolated from bronchial tissue, purchased from Procell, and subsequently cultured in specialized medium (Procell, cat#CM-H009). All cell lines were confirmed mycoplasma-free through monthly PCR testing.
RSV A2 and RSV ON1-GFP were described elsewhere (37). Recombinant live RSV carrying V127G, L141F, K399N, T400A, T400I, or F488L mutant of F protein was a kind gift from Gang Zou (Shanghai Ark Biopharmaceutical Co., Ltd, China). Recombinant live RSV carrying the K394R mutant of the F protein was a kind gift from Wencai Ye (Jinan University, China) (60, 64). All experiments involving live RSV were conducted in the biosafety level 2 (BSL-2) facilities in Guangzhou National Laboratory (GZNL). All viruses were propagated in HEp-2 cells and subsequently subjected to viral titer determination using a fluorescence focus assay (FFA), as previously described (37). Briefly, viral titrations were performed with 10-fold serial dilutions in HEp-2 cells. Twenty-four hours after inoculation, the cell supernatants were discarded, and the cells were fixed in 4% paraformaldehyde (PFA) for 15 min and then permeabilized with 0.3% Triton X-100 for 15 min at room temperature (RT). RSV was probed using anti-RSV-FITC (GeneTex, cat#GTX36375) diluted 1:100, over a 1 h incubation at RT. Fluorescent images were acquired with an Operetta CLS High-Content Screening System (PerkinElmer). Fluorescence dots were quantified and normalized to the total nuclear count using ImageJ analysis software (RRID: SCR_003070; National Institutes of Health, NIH).
Lonafarnib (cat#HY-15136), GS-5806 (cat#HY-16727), AK0529 (cat#HY-109142), RV521 (cat#HY-123475), and JNJ-53718678 (cat#HY-112180) were purchased from MedChemExpress. Compound 0179841 was synthesized as described below.
In vitro selection for RSV resistance to lonafarnib
To select for the development of drug resistance against lonafarnib, RSV A2 and RSV ON1-GFP strains were cultured in the presence of increasing concentrations of lonafarnib and passaged 18 times, respectively. Virus isolates recovered from culture at various passages were then characterized for their resistance to lonafarnib. To initiate passaging, HEp-2 cells were seeded in a 12-well plate at a density of 2 × 10^5^ cells per well, and both lonafarnib and virus were then added the following day. Lonafarnib was initially added at 250 nM, and the virus was added at an MOI of 0.1 per well. Two days post-infection, each well was scored for cytopathic effects (CPE) on an Operetta CLS High-Content Screening System (PerkinElmer), and 500 µL of the supernatant from the well with ~30% CPE was passaged to each well in the next culture plate. The remaining supernatant and cells were stored at −80°C until further analysis. The passage culture was set up in triplicate, and passaging was performed independently. Along with the cultures passaged with lonafarnib, RSV A2 and RSV ON1-GFP strains were passaged with 0.1% DMSO in three independent wells to serve as a passage control, respectively.
RSV full-genome sequence
The total RNA was extracted from the passaged supernatant and cell stocks using TRIzol LS reagent (Invitrogen, cat#10296028) according to the manufacturer’s instructions. Approximately 10 µg of the extracted RNA was then dispatched to Guangdong Magigen Biotechnology Co., Ltd for full-genome sequencing using the Novaseq technique. Ribosomal RNA was depleted from samples using the Ribo-off rRNA Depletion Kit (Vazyme Biotech Co., Ltd., cat#N406-01/02). Sequencing libraries were prepared with the ALFA-SEQ RNA Library Prep Kit II (Findrop Biosafety Technology [Guangzhou] Co., Ltd., cat#NRI002E) according to the manufacturer’s instructions. Constructed amplicon libraries are subjected to PE150 sequencing on the Illumina platform.
A workflow diagram of RSV single-nucleotide variant (SNV) calling is shown in Fig. S8. Briefly, raw sequencing data were processed using Fastp v0.23.2 to remove adapter sequences and low-quality reads. Reads with a quality score below 30 (Q30) and shorter than 36 bp were filtered out to ensure high-quality input for subsequent analyses (65). Filtered reads were aligned to the viral reference genome using BWA-MEM v0.7.17. The reference genome used was the RSV strain A2 or ON1, complete genome, available from the NCBI GenBank database (accession no. KT992094 or MW582528). PCR duplicates were marked and removed using Picard Tools v2.26.9. Sequencing depth was calculated using SAMtools v1.19.2 (66–68). To ensure comprehensive variant detection, we employed a multi-caller approach using five different variant calling algorithms: BCFtools v1.19 (Using htslib 1.19.1) (69), FreeBayes v1.3.6 (70), Mutect2 (GATK v4.2.5.0) (71), Strelka v2.9.10 (72), and VarScan v2.4.6 (73). Additionally, we utilized DeepVariant v1.6.0 (74), a deep learning-based variant caller, to complement our analysis. Amino acid changes resulting from the detected variants were annotated using SnpEff v5.2 (75). A custom annotation database was built using the RSV strain A2 or ON1 from the NCBI GenBank database (accession no. KT992094 or MW582528) to ensure accurate functional predictions. Frequency thresholds for reporting mutations were set at 5% for Illumina sequencing.
Evaluation of the in vitro antiviral activity
The inhibitory effects of compounds on wild-type RSV, passaged viruses, and recombinant RSV were evaluated in HEp-2 cells. The cells were seeded into 96-well plates at a density of 1.3 × 10^4^ cells per well for 20 h. The virus was then inoculated at a dose of 0.2 MOI per well, and a 3-fold dilution series of the indicated compounds was added in triplicate. Three days post-infection, the level of viral replication was quantitatively assessed by FFA, and EC_50_ values were derived by fitting a nonlinear regression curve to the data in GraphPad Prism 8.0 software (RRID: SCR_002798, Graphpad Software Inc.). EC_90_ values of lonafarnib for recombinant RSV were calculated by the formula (EC_90_ = 9^1/HillSlope*EC_50_).
The inhibitory effects of compounds on RSV ON1-GFP were evaluated in HBEC. Specifically, 1.3 × 10^4^ cells per well were seeded into 96-well plates, infected with RSV ON1-GFP at an MOI of 2, and subsequently incubated with either lonafarnib or compound 0179841. At 72 h post-infection, the infected cells were visualized using an EVOS M5000 Cell Imaging System (Thermo Fisher Scientific). The GFP intensity was quantified in five randomly selected fields per experiment using ImageJ analysis software (RRID: SCR_003070; National Institutes of Health, NIH).
Cytotoxicity assay
HEp-2 cells (1.3 × 10^4^) were seeded in 96-well plates for 20 h. The indicated compounds in a 3-fold dilution series were added to the cells, and three wells were performed in parallel. After 72 h, the cells were incubated with DMEM containing 10% FBS and CCK8 reagent (Beyotime Biotechnology, cat#C0038) for 30 min at 37°C. The absorbance at 450 nm and 600 nm was read using the PerkinElmer EnSight reader.
Cell–cell fusion assay
Cell–cell fusion assays were performed using a split-GFP system (GFP1-10 and GFP11) (76). Briefly, HEK293T cells stably expressing GFP1-10 were transfected with 1,000 ng of plasmids encoding either wild-type F or F mutants containing various amino acid substitutions. Alternatively, the cells were infected with RSV and treated with 0.1% DMSO or the indicated compounds at concentrations of 5, 1, and 0.5 µM. Then, the transfected or infected cells were co-cultured with HEK293T cells stably expressing GFP11 at a 1:1 ratio (8 × 10^4^ cells/well) for 24 h in a 96-well plate. Images were acquired per well on an Operetta CLS High-Content Screening System (PerkinElmer). The GFP amounts were quantified using ImageJ analysis software (RRID: SCR_003070; National Institutes of Health, NIH).
Protein production
The cDNAs encoding prefusion-stabilized RSV F variant DS-Cav1 were cloned into the pcDNA3.1 expression vector as previously described (19, 37). The DS-Cav1 mutants were constructed using a site-directed mutagenesis kit. The DS-Cav1 and its mutants were expressed in HEK293F cells with polyethylenimine (PEI, Polysciences Inc., cat#24765) as previously described (37). Briefly, the cell supernatant was harvested, centrifuged, and filtered at 4 days post-transfection. The complex was initially purified with Ni^2+^-NTA resin (Cytiva, cat#17531801) using an elution buffer (1 × PBS pH 7.4, 500 mM imidazole). The proteins were further purified using a Superose 6 10/300 Gl gel filtration column (Cytiva, cat#17517201) with running buffer consisting of 1 × PBS pH 7.4, then concentrated to about 1 mg/mL.
SPR analysis
SPR assays were performed as previously described (37). Briefly, the interaction between RSV F proteins and lonafarnib was detected using a Biacore 8K (GE Healthcare) at 25°C in a multi-cycle mode. RSV F proteins were immobilized on a Series S Sensor chip CM7 (Cytiva, cat#29147020). Lonafarnib with concentrations of 0.78125, 1.5625, 3.125, 6.25, and 12.5 µM was prepared in buffer containing 10 mM PBS pH 7.4, 0.05% Tween 20, and 5% DMSO when testing interactions with RSV F proteins. The equilibrium dissociation constants (KD) for each pair of interactions were calculated using the Biacore 8K evaluation software (Cytiva).
Molecular dynamics simulation
The molecular dynamics simulations were performed on a SingleParticle S23TL24 workstation configured with 2× Intel Xeon Gold Platinum 8352 v Processors, 512 GB of RAM, and 4× Nvidia RTX 4090. System preparation was conducted using the Protein Preparation Wizard (RRID: SCR_016749) in Schrödinger Maestro (Schrödinger Release 2020-1, Schrödinger, LLC). The simulation systems were solvated using the SPC water model and neutralized with a 150 mM NaCl buffer. Each system, containing trimeric RSV F proteins and lonafarnib, comprised approximately 125,000 atoms within an orthorhombic 10 × 10 × 10 Å^3^ cubic box. Production simulations were carried out in the NPT ensemble, maintaining a temperature of 300 K and a pressure of 1.01325 bar. The simulations, executed with Desmond (RRID: SCR_014575) using the OPLS4 force field, ran for 500 ns each, with data recorded every 500 ps. The structural representations shown in the figures were rendered using PyMOL v.3.0.4 (RRID: SCR_000305; Schrödinger, LLC).
Molecular docking
Protein structures of RSV F (PDB: 8KG5) and CRBD (PDB: 6BN7) were prepared for docking by retaining structural monomers, correcting structures, adding hydrogen atoms, and optimizing side chains. The resulting protein complex models were imported into MOE v2022.02 (RRID: SCR_014882) software via HDOCK v1.1 (RRID: SCR_024799). The MOE Dock module was used to dock compound 0179841 with the protein complex cereblon and RSV F. The best binding poses were identified based on geometric and energy matching, yielding a binding score of −11.38. A more negative score indicates stronger binding affinity, and the selected pose was used for interaction analysis.
Synthesis of compounds
tert-butyl
3-(2-(2-(2-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)ethoxy)ethoxy)ethoxy)propanoate (2)
In total, 2-(2,6-Dioxopiperidin-3-yl)−4-fluoroisoindoline-1,3-dione (550 mg, 1.98 mmol) and DIPEA (1.1 mL, 6.54 mmol) were added to a solution of compound 1 (500 mg, 1.8 mmol) in DMSO (8 mL). The mixture was stirred at 90°C for 2.5 h. After being cooled to RT, the mixture was diluted with H_2_O and extracted with ethyl acetate (EA), the organic layer was washed with brine and dried over Na_2_SO_4_. The solvent was evaporated under reduced pressure, then purified by flash column, eluting with a gradient of 0%–2.5% MeOH in Dichloromethane (DCM) to give compound two as an emerald green oil (180 mg, yield 19%). HRMS (ESI) m/z: calculated for C_26_H_35_N_3_O_9_ [M + H]^+^ 534.2446, found 534.2442.
3-(2-(2-(2-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)ethoxy)ethoxy)ethoxy)propanoic acid (3)
TFA (3 mL, 41 mmol) at 0°C was added to a solution of compound 2 (180 mg, 0.35 mmol) in DCM (5 mL). After the addition, the resulting mixture was stirred for 2 h while allowing the temperature to rise slowly to RT. The mixture was spun dry to remove TFA and then purified by flash column, eluting with a gradient of 0%–2.5% MeOH in DCM to give compound three as a light-yellow oil (70 mg, yield 42%), which was used for the next step directly. HRMS (ESI) m/z: calculated for C_22_H_27_N_3_O_9_ [M + H]^+^ 478.1820, found 478.1811.
4-((2-(2-(2-(3-(4-(2-(4-((R)−3,10-dibromo-8-chloro-6,11-dihydro-5H-benzo[5,6]cyclohepta[1,2-b]pyridin-11-yl)piperidin-1-yl)−2-oxoethyl)piperidin-1-yl)−3-oxopropoxy)ethoxy)ethoxy)ethyl)amino)−2-(2,6-dioxopiperidin-3-yl)isoindoline-1,3-dione (0179841)
To a solution of compound 3 (70 mg, 0.151 mmol) in DMF (3 mL) was added (R)−1-(4-(3,10-dibromo-8-chloro-6,11-dihydro-5*H-*benzo[5,6]cyclohepta[1,2-b]pyridin-11-yl)piperidin-1-yl)−2-(piperidin-4-yl)ethan-1-one (85 mg, 0.138 mmol), then added HATU (83 mg, 0.302 mmol), DIPEA (95 µL, 0.453 mmol). The mixture was stirred for 3 h, then diluted with H_2_O and extracted with EA (20 mL), the organic layer was washed with brine and dried over Na_2_SO_4_. The solvent was evaporated under reduced pressure. The residue was purified by flash column chromatography, eluting with a gradient of 0%–3.3% MeOH in DCM to give compound 0179841 as a light yellow solid (70 mg, yield 46%). HPLC: 98% (t_R_ = 14.319 min); ^1^H NMR (300 MHz, CDCl_3_) δ = 8.86 (s, 1H), 8.45 (s, 1H), 7.56 (s, 1H), 7.54–7.43 (m, 2H), 7.17–7.05 (m, 2H), 6.91 (d, J = 8.4 Hz, 1H), 6.50 (s, 1H), 4.91 (d, J = 10.1 Hz, 2H), 4.59 (d, J = 12.7 Hz, 2H), 3.89–3.76 (m, 4H), 3.76–3.55 (m, 12H), 3.46 (s, 2H), 3.27 (d, J = 17.8 Hz, 1H), 3.01 (s, 2H), 2.93–2.70 (m, 6H), 2.61 (q, J = 10.5 Hz, 3H), 2.38 (s, 2H), 2.15 (d, J = 28.4 Hz, 4H), 1.83 (d, J = 12.9 Hz, 1H), 1.73 (d, J = 13.2 Hz, 1H), 1.38 (d, J = 16.7 Hz, 2H), and 1.12 (s, 2H); HRMS (ESI) m/z: calculated for C_48_H_55_Br_2_ClN_6_O_9_ [M + H]^+^ 1053.2159, found 1053.2100.
Western blot
HEK293T cells were seeded in 12-well plates (80% confluence) 1 day before F plasmids transfection and compound treatment. On the day of transfection, the cell culture medium was first changed, and then, the cells were transfected with 2 µg of F plasmids. In the assays for validating the degradation of F, 10 mM DMSO stocks of the indicated compounds were added into the medium (working concentrations = 1 µM, 0.1% DMSO) for the indicated periods of time post-transfection. Cells were scraped, washed twice with PBS, and lysed with cell lysis buffer (Beyotime Biotechnology, cat#P0013), supplemented with protease inhibitor PMSF (Beyotime Biotechnology, cat#ST505) for 30 min on ice. Cell lysates were clarified by centrifugation at 10,000 × g for 10 min at 4°C. Protein concentrations were determined using Enhanced BCA Protein Assay Kit (Beyotime Biotechnology, cat#P0009). 5 × SDS PAGE Sample Loading Buffer (Beyotime Biotechnology, cat#P0015L) was added to the samples and boiled at 100°C for 10 min. The equal concentrations of protein samples were separated by SDS-PAGE, and proteins were transferred from gels to PVDF membranes (Merck, cat#ISEQ00010). The membranes were incubated in 5% wt/vol milk in 0.1% Tween-20/PBS at RT for 1 h, incubated with the indicated primary antibodies (mouse anti-RSV F antibody, Abcam, cat#ab43812, RRID: AB_777676; mouse anti-β-actin antibody, Proteintech, cat#66009-1-Ig, RRID: AB_2687938) diluted in 0.1% Tween-20/PBS at 4°C overnight and incubated with the indicated secondary antibodies (Peroxidase AffiniPure Goat Anti-Mouse IgG, Jackson Immuno Research, cat#115-035-146, RRID:AB_2307392) diluted in 0.1% Tween-20/PBS at RT for 1 h. The membranes were washed by 0.1% Tween-20/PBS after each incubation. Signal bands were exposed and quantified using the FluorChem HD2 system (Alpha Innotech).
Antiviral drug assay using lung airway organoids
HBECs-derived airway organoids were kindly provided by Ning Ma (Guangzhou National Laboratory, China). In brief, HBECs were initially cultured in PneumaCult-Ex Plus Medium (STEMCELL Technologies, cat#05040), then seeded at 2 × 10⁵ cells/well into anti-adherence rinsing solution (STEMCELL Technologies, cat#07010)-treated AggreWell400 24-well plates (STEMCELL Technologies, cat#34415). They were maintained in PneumaCult Apical-Out Airway Organoid Medium (STEMCELL Technologies, cat#100-0620) for 6 days. Subsequently, the cells were transferred to similarly treated flat-bottom 24-well plates and cultured in the same medium for an additional 9 days.
The airway organoids were infected with RSV ON1-GFP at an MOI of 1 for 4 h. After removal of the inoculum, the organoids were washed twice with PBS, and the infected organoids were cultured in medium containing 0.1% DMSO or serially diluted compound 0179841. At 96 h post-infection, cell-free medium was harvested, and a TCID_50_ assay was performed to determine the viral titer. The organoids were fixed in parallel in 4% PFA, permeabilized with 0.5% Triton X-100 (1 h), blocked with 5% BSA (Macklin, cat#B824162; 2 h), and incubated overnight at 4°C with primary antibodies: Goat anti-Human p63/TP73L polyclonal antibody (R&D Systems, cat#AF1916), Rabbit anti-Human Mucin 5AC polyclonal antibody (Abways, cat#CY6826), and Mouse anti-acetylated tubulin monoclonal antibody (Sigma-Aldrich, cat#T7451). After being washed three times with PBS, the samples were stained with Donkey anti-Goat IgG (H + L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 633 (Thermo Fisher Scientific, cat#A-21082; 1 hour, RT), Donkey anti-Rabbit IgG (H + L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor 568 (Thermo Fisher Scientific, cat#A-10042; 1 h, RT), and Donkey anti-Mouse IgG (H + L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor 568 (Thermo Fisher Scientific, cat#A-10037; 1 h, RT), followed by Hoechst 33342 nuclear counterstain (Thermo Fisher Scientific, cat#62249). After being washed three times with PBS, images were acquired on a Nikon A1 confocal microscope (Nikon). The GFP fluorescence intensity was quantified using ImageJ analysis software (RRID: SCR_003070; National Institutes of Health, NIH).
Statistical analysis
The data in the figures represent means ± SDs and were analyzed using GraphPad Prism 8.0 software (RRID: SCR_002798, Graphpad Software Inc.). The linkage relationship between mutations was analyzed using a χ^2^ test for independence. For the cell–cell fusion assay, statistical comparisons between different groups were performed using the non-parametric Mann-Whitney test, combining data from several experiments. The differences between the experimental and control groups were determined by Student’s t-test involving two groups or one-way ANOVA, followed by Dunnett’s post hoc test for multiple group comparisons. P‐values were calculated, and statistical significance was expressed as *P < 0.05, **P < 0.01, ***P < 0.001; ns, not significant. All experiments were performed with at least three biological replicates. The details of the statistical analyses were described in the figure legends.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Li Y, Wang X, Blau DM, Caballero MT, Feikin DR, Gill CJ, Madhi SA, Omer SB, Simões EAF, Campbell H, et al.. 2022. Global, regional, and national disease burden estimates of acute lower respiratory infections due to respiratory syncytial virus in children younger than 5 years in 2019: a systematic analysis. The Lancet 399:2047–2064. doi:10.1016/S 0140-6736(22)00478-0PMC 761357435598608 · doi ↗ · pubmed ↗
- 2Nguyen-Van-Tam JS, O’Leary M, Martin ET, Heijnen E, Callendret B, Fleischhackl R, Comeaux C, Tran TMP, Weber K. 2022. Burden of respiratory syncytial virus infection in older and high-risk adults: a systematic review and meta-analysis of the evidence from developed countries. Eur Respir Rev 31:220105. doi:10.1183/16000617.0105-202236384703 PMC 9724807 · doi ↗ · pubmed ↗
- 3Papi A, Ison MG, Langley JM, Lee D-G, Leroux-Roels I, Martinon-Torres F, Schwarz TF, van Zyl-Smit RN, Campora L, Dezutter N, de Schrevel N, Fissette L, David M-P, Van der Wielen M, Kostanyan L, Hulstrøm V. 2023. Respiratory syncytial virus prefusion f protein vaccine in older adults. N Engl J Med 388:595–608. doi:10.1056/NEJ Moa 220960436791160 · doi ↗ · pubmed ↗
- 4Walsh EE, Pérez Marc G, Zareba AM, Falsey AR, Jiang Q, Patton M, Polack FP, Llapur C, Doreski PA, Ilangovan K, et al.. 2023. Efficacy and safety of a bivalent RSV prefusion F vaccine in older adults. N Engl J Med 388:1465–1477. doi:10.1056/NEJ Moa 221383637018468 · doi ↗ · pubmed ↗
- 5Wilson E, Goswami J, Baqui AH, Doreski PA, Perez-Marc G, Zaman K, Monroy J, Duncan CJA, Ujiie M, Rämet M, et al.. 2023. Efficacy and safety of an m RNA-based RSV Pre F vaccine in older adults. N Engl J Med 389:2233–2244. doi:10.1056/NEJ Moa 230707938091530 · doi ↗ · pubmed ↗
- 6The I Mpact-RSV Study Group. 1998. Palivizumab, a humanized respiratory syncytial virus monoclonal antibody, reduces hospitalization from respiratory syncytial virus infection in high-risk infants. Pediatrics 102:531–537. doi:10.1542/peds.102.3.5319724660 · doi ↗ · pubmed ↗
- 7Jones JM, Fleming-Dutra KE, Prill MM, Roper LE, Brooks O, Sánchez PJ, Kotton CN, Mahon BE, Meyer S, Long SS, Mc Morrow ML. 2023. Use of nirsevimab for the prevention of respiratory syncytial virus disease among infants and young children: recommendations of the advisory committee on immunization practices - United States, 2023. MMWR Morb Mortal Wkly Rep 72:920–925. doi:10.15585/mmwr.mm 7234 a 437616235 PMC 10468217 · doi ↗ · pubmed ↗
- 8Anastassopoulou C, Medić S, Ferous S, Boufidou F, Tsakris A. 2025. Development, current status, and remaining challenges for respiratory syncytial virus vaccines. Vaccines (Basel) 13:97. doi:10.3390/vaccines 1302009740006644 PMC 11860200 · doi ↗ · pubmed ↗