A Pathway to High Quality Clinical Trials in IgA Vasculitis Nephritis: Meeting Proceedings From a Multiprofessional International Collaborative Workshop
Louise Oni, Rona Smith, Seza Ozen, Chloe Williams, Elin Davies, Charlotte King, Paul Brogan, Mark Haas, Jonathan Barratt, Jeffrey Hafkin, Despina Eleftheriou, Karuna Keat, EMD Smith, Wen Ding, Chee Cheung, Caroline Platt, Evangéline Pillebout, Andrew Chetwynd, Areefa Alladin

TL;DR
Experts from around the world met to identify barriers and create a plan for better clinical trials in IgA vasculitis nephritis, aiming to improve patient outcomes.
Contribution
The paper presents a collaborative action plan to develop high-quality clinical trials for IgA vasculitis nephritis through international expert consensus.
Findings
There is sufficient similarity between adult and pediatric IgAV-N to justify combined clinical trials.
Patients at highest risk of kidney failure should be prioritized in initial trials.
Key considerations include diagnostic classification, age-inclusive trial design, and patient-reported outcomes.
Abstract
IgA vasculitis (IgAV) is an autoimmune disease that affects the small vessels of the skin, joints, gastrointestinal (GI) tract, and kidneys. In the long term, IgAV associated with nephritis (IgAV-N) can progress to kidney failure. Evidence-based clinical studies of IgAV-N are few, leading to huge variations in treatment approaches and suboptimal outcomes. The wealth of emerging efficacious treatments for IgA nephrology brings new opportunities to this disease. The aim of this report is to describe the proceedings of a multiprofessional collaborative workshop convened to identify the barriers to developing high quality evidence for patients with IgAV-N. A multiprofessional group consisting of 53 attendees from 13 countries met. The meeting was represented by a variety of professional backgrounds, including lay attendees, with different levels of expertise (32% professors and 19%…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
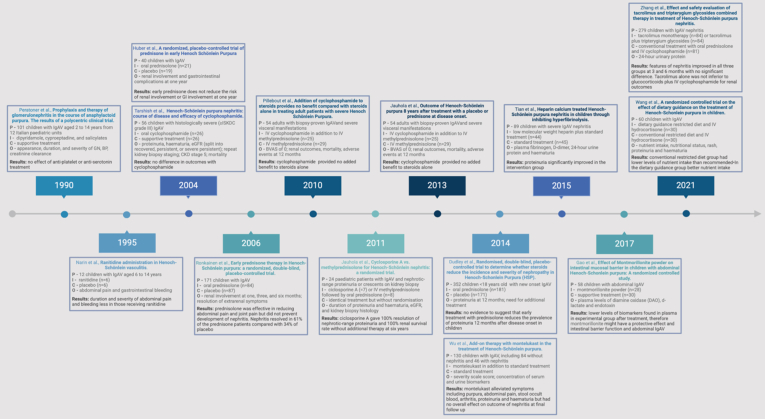 Figure 1
Figure 1Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsVasculitis and related conditions · Renal Diseases and Glomerulopathies · Systemic Sclerosis and Related Diseases
Introduction
IgAV is a small vessel autoimmune vasculitis that is mediated by perivascular deposition of immune complexes containing galactose-deficient-IgA1. Although IgAV is a rare disease, it is the most common form of vasculitis seen in the pediatric population with an incidence of about 150 to 200 cases per million population and a leading cause for a pediatric nephrologist to conduct a kidney biopsy.1^,^2 In adults, it is far less common with an incidence of about 1 case per million population and there remains major gaps in identifying the true incidence of adult onset IgAV, with studies skewed toward those with major organ involvement when compared with childhood onset cohorts.2 For children, there are internationally recognized EULAR/PRINTO/PReS classification criteria3 and the European IgAV study group are currently developing adult consensus criteria. The etiology of this disease is multifactorial and geographical variation plus environmental triggers have been described.4 Polygenic risk variants are also reported, such as those implicated with human leucocyte antigen, cytokine mediator pathways, and the complement pathway.5 IgAV can be triggered by the stimulation of the immune system through acute infection or vaccinations; and mostly in adults, it may be secondary to malignancy, liver disease, or drug induced.6
Histologically, IgAV is identified as a small vessel leukocytoclastic vasculitis and the disease typically involves 4 organs. These organs include the skin, particularly the postcapillary venules; the joints; the GI tract; and the kidneys. In most patients, the striking lower limb cutaneous skin vasculitis is the initial symptom, and this prompts a medical review leading to a diagnosis. In the short term, skin manifestations, joint pain, and GI involvement predominate; however, a third of patients will subsequently develop nephritis (known as IgAV-N) occurring days, weeks, or months after the initial presentation.7 Particularly in children, the disease is a monophasic illness, that can spontaneously remit with no long-term sequalae and a positive outcome. Spontaneous remission in cases of biopsy-proven IgAV-N is less common and the condition in adolescents and adults may be more persistent.8^,^9 In the long term, kidney involvement contributes the most to morbidity, with nephritis observed in about 30% of patients during the acute phase.4^,^10 Limited observational data demonstrates that the kidney outcomes are not benign with about 20% of patients with biopsy-proven IgAV-N having significant levels of proteinuria 10 years after disease onset.11 The kidney outcomes correlate with age, with 1% to 3% of all children presenting with IgAV being at risk of kidney failure and this increases to 24% in the elderly.12 Failure to achieve complete remission of proteinuria, like other forms of glomerulonephritis, is associated with chronic kidney disease progression.13 Furthermore, some patients may relapse in adulthood, or evolve into IgAN with single organ involvement of only the kidney; indeed, IgAN and IgAV-N cannot be distinguished from each other on kidney biopsy. Approximately 12% of patients with IgAV-N can develop recurrence of IgA-related disease in kidney transplant, emphasizing the importance of early intervention to prevent the journey to chronic kidney disease.14
Patients with IgAV-N at high risk of kidney disease progression have been characterized. In a study evaluating the clinical features and kidney biopsy findings of 262 children and 99 adults, the high risk patients (defined as a risk of ≥30% decline in estimated glomerular filtration rate or developing end-stage kidney failure) had more noncutaneous manifestations, a lower estimated glomerular filtration rate at the time of kidney biopsy, were aged ≤ 18 years, or had histological presence of endocapillary hypercellularity.15 Similar findings have been reported in meta-analysis studies.10 Although defining high risk may be achievable, there remains a lack of objective measure to quantify overall disease activity because the Birmingham vasculitis activity score, and its pediatric equivalent, have limited validation in this specific form of vasculitis.16^,^17
For the management of nephritis, multicenter cohort studies report the frequent use of broad-spectrum immunosuppression, predominantly the use of corticosteroids and mycophenolate mofetil; however, calcineurin inhibitors and B-cell depletion have been used in both children and adults.15^,^18^,^19 Previous clinical trials have been small and heterogenous (Figure 1); however, there is perhaps a trend toward more targeted therapies, and an open label clinical trial in 54 adults with biopsy-proven IgAV-N demonstrated no benefit from the addition of cyclophosphamide to corticosteroids on improving Birmingham vasculitis activity score scores.20 As a result, Cochrane reviews have repeatedly been unable to support evidence-based recommendations for IgAV,21 which have led to expert consensus-based approaches, including the European Single Hub and Access Point for Paediatric Rheumatology initiative.22 In patients with suspected active nephritis, the indications for when to perform a kidney biopsy have been aligned in the International Pediatric Nephrology Association guidelines and the Kidney Diseases: Improving Global Outcomes glomerular diseases executive summary.22, 23, 24Figure 1A timeline illustrating the clinical trials that have been conducted in patients with IgA vasculitis since 1990. CKD, chronic kidney disease; eGFR, estimated glomerular filtration rate; IgAV, IgA vasculitis.
There is a major unmet need for evidence-based treatments in IgAV-N and it is the leading cause of morbidity. Considering the recognition that the disease pathogenesis of IgAV-N shares major similarities with IgAN,25 such as the involvement of circulating galactose-deficient IgA1 and histological classification scores validated for IgAN perform relatively well in IgAV-N,15^,^26^,^27 this may provide the opportunity to reposition treatments because of the wealth of active clinical trials in IgAN.28 Furthermore, the visible rash seen in patients with IgAV may eventually provide a window of opportunity to intervene early, a situation not seen in many other forms of glomerulonephritis; meaning complete prevention of chronic kidney disease may even be feasible. Because of the complexities of conducting clinical trials in rare, clinically heterogeneous diseases, especially those predominating in children, changing the treatment paradigm for kidney disease secondary to IgAV requires a highly collaborative and age-inclusive approach.
Aim
The aim of this paper was to report the outcome of an international multiprofessional collaborative workshop to develop a pathway to drive high-quality clinical trials for patients with IgAV-N.
Methodology
Objectives
The objectives of the event were as follows:
- 1.To determine the similarities between pediatric and adult-onset disease.
- 2.To explore treatments in other glomerular or vasculitic conditions that could be evaluated in IgAV-N.
- 3.To agree on the subgroup of patients with IgAV-N at high risk of progression to kidney failure.
- 4.To determine whether extrapolation, bridging biomarkers, or novel trial designs could be considered.
- 5.To discuss inclusive trial designs to capture all ages of patients who experience this disease.
- 6.To identify suitable outcome measures for treatment efficacy.
Participants
Participants were invited to attend through existing international networks that included vasculitis networks (UK Ireland Vasculitis Society; European IgA Vasculitis Group (i.e., EUGAVAS study group); Pediatric Rheumatology European Society vasculitis working party, Australia and New Zealand Vasculitis Society, glomerulonephritis groups (UK Glomerulonephritis Clinical Studies Group), pediatric groups (British Association for Paediatric Nephrology, European Paediatric Nephrology Association Glomerular Diseases Working Group, Paediatric Rheumatology European Society) and IgA nephropathy collaborations (International IgA Nephropathy Research Network, European Academy of Dermatology and Venereology, Task Force vasculitis and vasculopathy). Expressions of interest were open for a period of 3 months and then places were confirmed. Of the confirmed attendees, 12 participants, representing diverse personal and professional characteristics, were invited to present short summaries aligned with their area of expertise as part of the agenda. Patients were represented by specialist charity partners. All participants received reimbursement for travel, accommodation, and subsidence. Colleagues who were unable to attend the event in person were encouraged to engage and contribute before and after the event using email correspondence or virtual meetings.
Setting
The group of multiprofessional experts convened on February 13 and 14, 2025 at Alder Hey Children’s NHS Foundation Trust Hospital in Liverpool, UK.
Agenda
The agenda was divided into 2 sections with brief evidence-based presentations, interspersed with discussion. The agenda covered the natural history of the disease in both children and adults to inform similarities and opportunities to intervene during the disease course. The study population, interventions, comparators, primary and secondary outcomes, adaptations required to be age inclusive, feasibility, and power were discussed. Biobanking and additional exploratory studies were included together with the importance of patient-reported outcome measures. The event agenda concluded with exploring funding opportunities and confirmation of action points.
Data Extraction
Using the aims and objectives within the framework of the agenda, key themes were extracted from the event, an action plan of critical evidence gaps was developed and proposed clinical trial ideas were generated.
Funding and Approvals
The meeting was funded as part of the LifeArc-KRUK Translational Centre for Rare Kidney Disease project (LifeArc is a charity registered in England and Wales under no. 1015243 and in Scotland under no. SC037861; grant no. 10749) in addition to a voluntary donation provided as an independent educational grant from CSL Vifor. No formal approvals were required for this project.
Results
Participants and Introduction
The 2-day event included 53 attendees, representing 13 countries across 5 continents. A total of 39 participants (74%) attended from the host country, UK and 14 (26%) were from other countries. The countries represented included UK, Ireland, USA, Canada, Australia, Turkey, France, Italy, Germany, The Netherlands, Spain, Belgium, Croatia, Romania, Hungary, Saudi Arabia, and Brazil. The professions represented included pediatric nephrology consultants, pediatric rheumatology consultants, pediatric vasculitis consultants, adult nephrology consultants, pediatric trainees, nephrology trainees, histopathologists, immunologists, industry colleagues, trialists, scientists, a dermatologist, a patient-reported outcome measurement expert, an operational manager, an administrator, and a statistician. The participants were of varying stages of their professional careers with 17 professors and 10 midcareer doctors.
Patient Involvement
Patient representatives stated, “It is shocking that nothing has changed in 20 years” and their engagement as patient partners was highlighted. The inclusion of patient-reported outcome measures to capture the burden of symptoms relevant to this disease was included in the discussion, using the selection of patient-reported outcome measures validated to regulatory standards, such as symptomatic edema24 and those capturing vasculitic features.29 Notably, the need for parent- or caregiver-reported measures to capture symptom and functional impacts among young children will be needed to ensure this group is captured. Patient involvement is imperative to drive the importance of evidence in this disease because the current narrative emphasizes that this disease is fully self-resolving and only occurs in children. A patient-led awareness campaign may help to change this perception.
Childhood and Adult-onset IgAV Disease
The threshold where professionals would regard the degree of similarity in the disease and the expected response to treatment between 2 groups (children and adults) was discussed. There is no specific threshold for sufficient similarity; however, 70% is often considered adequate when using modified Delphi techniques.30 The experts felt that there was sufficient similarity related to the organs involved; and specifically, skin manifestations across the pediatric and adult populations. The group agreed that the pathophysiology, and therefore the mechanisms of action of treatments, are likely to be sufficiently similar for all ages, while appreciating that diagnosis in adults may be more challenging in the absence of consensus classification criteria. The histopathological features in IgAV-N across children and adults were believed to be sufficiently similar.15 However, additional clinical features, the disease course, and kidney failure outcomes were not deemed to be sufficiently similar between the age groups, and these elements would need to be considered when designing a trial.
IgAV-N and Other Glomerulonephritides
The kidney histology in patients with IgAV-N when compared with the histology of IgAN were deemed to be highly similar. It was noted that endocapillary hypercellularity is the most important histologic feature with regard to clinical outcomes in IgAV-N.15^,^31 Therefore, the Oxford MEST-C classification, developed and validated for IgAN in adults and children, appears the most suitable for histological scoring in IgAV-N when compared with the International Study of Kidney Diseases in Children classification, which does not include endocapillary hypercellularity.31 The use of the Oxford MEST-C scoring system in this condition has the added benefit of providing consistency with trial protocols in IgAN. Glomerular deposits of complement C3 and their colocalization with IgA were recognized to correlate with disease severity in both IgAN and IgAV-N.32 Therefore, drugs targeting complement pathways and especially the alternative pathway, were felt to be promising because of the biological similarities and proven efficacy in other forms of glomerulonephritis. Patients going into spontaneous remission was expressed as a trial design challenge. Screening patients from the time of diagnosis and ensuring ≥2 consecutive elevated urine proteinuria measurements before entry may be a feasible screening method to exclude those who may naturally remit early. It was also recognized that fewer patients remit by the time they undergo a kidney biopsy and using this high-risk group to begin to accelerate trials in this disease may be the most successful option. Enriching the natural history data to better describe IgAV-N would inform the precise population and guide clinical trial design, and hopefully act as an incentive to support industry collaborations, as was demonstrated by the impactful long-term data obtained in IgAN.33 Observational data so far suggests that the current standard-of-care for immunosuppressive treatments is most frequently corticosteroids and mycophenolate mofetil.18 Other considerations included establishing the long-term cost implications of IgAV-N, which largely affects younger individuals and how chronic kidney disease progression is costly over the life course. A formal health economic analysis would justify the treatment costs and help secure successful implementation of treatments into clinical care following evidence generation.
Defining the Population at High Risk of Kidney Progression Because of IgAV
The working group agreed that gaining evidence to guide the management of nephritis was a priority for patients with this condition, with active nephritis being defined by increased proteinuria, with or without impaired kidney function and/or biopsy-proven inflammation. In terms of other organ involvement, GI involvement was felt to be the second priority, followed by cutaneous involvement, then joint involvement, which is often self-limiting. With regard to early markers of kidney disease, isolated microscopic hematuria was felt to be of unknown significance. Reducing the variability on when to perform a kidney biopsy was highlighted, with attendees suggesting that a urine protein-to-creatinine ratio value ranging between 100 and 500 mg/mmol Cr would be suitable inclusion criterion. The recent International Pediatric Nephrology Association guidelines, which outline kidney biopsy indications with a urine protein-to-creatinine ratio > 200 mg/mmol Cr and/or a reduced estimated glomerular filtration rate < 90 ml/min per 1.73 m^2^ attributable to active disease, moderate proteinuria of 100 to 200 mg/mmol Cr if present for > 2 weeks and urine protein-to-creatinine ratio is 20 to 100 mg/mmol Cr for 4 to 12 weeks, if there has been no improvement after renin angiotensin-aldosterone system inhibition, may act as a suitable benchmark.24
Attendees acknowledged that disease scoring using the Birmingham vasculitis activity score and pediatric equivalent has previously formed part of composite outcome measure in trials for other forms of vasculitis that have aided drug licensing approvals and could be considered. There was recognition of the need to validate these tools, or adjust them to be more disease-specific, because the validation of Birmingham vasculitis activity score (version 3) only included 10 adults with IgAV (3.2%)34 and only 1 child (1.6%) was included in the validation of the pediatric equivalent despite it being the most common form of vasculitis in children.17 Preliminary creation of a disease-specific scoring tool “The IgA-VAS,” to support better phenotyping and/or objectively measure outcomes, was presented and this would require further refinement and validation in a fully representative cohort. This could be achieved by forming an international expert consensus panel and validation patient cohorts to mature the preliminary tool.
Extrapolation, Bridging Biomarkers, Scientific Markers, and Novel Trial Designs
Overall, a phase 2 proof of concept study would ensure feasibility of conducting trials in this population to prepare for confirmatory phase 3 clinical trials. This may be best achieved by using the current regulatory approved end points for nephrology trials, including proteinuria reduction and change in estimated glomerular filtration rate as primary outcome measures. A protocol would require safety and drug kinetics that are relevant for all ages. Generating label-enabling data were reported to be an incentive for industry partners.
Novel trial designs, including the use of Bayesian approaches to incorporate external data were discussed. A multiarm platform trial design, that consists of a master protocol where multiple trial interventions can be evaluated more efficiently, was seen as a superior way to establish a perpetual framework to evaluate treatments within a shorter time frame and to enable evaluation of the wealth of treatments evolving for IgAN.35 The trial templates and progression of evidence used for other rare kidney diseases, such as C3 glomerulopathy and IgAN, could provide rich learning; and exemplars integrating genetic stratification are emerging.36^,^37 Consideration of how to define and manage a disease flare during a trial would need to be included. The group felt that combining adult patients with IgAV-N into IgAN studies, perhaps using an umbrella design, was beginning to be used in a few studies; however, there were concerns about recruitment numbers for adults with IgAV-N and whether it may hinder the progress being made in IgAN. In contrast, in the pediatric population, where many of the agents have not yet been evaluated in children with IgAN, it was felt that there could be an opportunity to merge IgAN and IgAV-N although this would need further consideration.
Considerations to Ensure Feasibility of a Clinical Trial in IgAV
The group agreed that the medications needed to be in a suitable formulation for administration to patients of all ages. This means that some agents, such as the targeted-release formulation of budesonide (nefecon), may not be appropriate for evaluation across all ages because they are modelled on the size of an adult GI tract. It was felt that regulators would need to be satisfied that the 2 diseases, IgAV-N and IgAN, were sufficiently scientifically similar to justify the evaluation of similar treatments. Ongoing research to explore the biological similarities is therefore encouraged and a subsequent survey after this event has demonstrated that 76.5% of the Renal Pathology Society members would prefer to report a kidney biopsy for IgAV-N using the Oxford MEST-C classification. Potential future therapeutic agents were discussed, and these are presented in detail in Table 1.38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48, 49, 50, 51, 52, 53, 54, 55, 56, 57, 58, 59, 60, 61, 62, 63, 64, 65, 66, 67, 68, 69 There were collective desires to investigate novel biomarkers, such as galactose-deficient IgA or B-cell repertoire, to better stratify patients as exploratory components in a trial.Table 1A list of potential pipeline therapeutic agents from the literature that could be considered in IgAV-N, grouped according to mechanism of actionAgentTherapeutic subclassSupporting comments and evidence in IgAV, IgAN or other forms of vasculitisTargeting B-cell pathwaysRituximabDepletes CD20 cells, chimeric formulationCase series in IgAV19^,^38^,^39RIGA trial underway in adults with IgAV.AAV induction and maintenance.40ObinutuzumabDepletes CD20 cells, fully humanized formulationDeeper B-cell depletion than rituximab, evaluated in several diseases including lupus nephritis,41 membranous nephropathy,42 ANCA-associated vasculitis43 and childhood nephrotic syndrome.44SibeprenlimabInhibits APRILENVISION phase 2 trial with VISIONARY phase 3 trial in IgAN awaiting completion.45ZigakibartInhibits APRILBEYOND phase 3 trial in IgAN awaiting outcome.46AtaciceptRecombinant fusion protein binds BAFF and APRILORIGIN Phase 2b trial in IgAN reduced proteinuria with a positive open label extension study and phase 3 awaiting completion.47^,^48 PIONEER trial is underway and includes adults and adolescents with a range of glomerular diseases including IgAV-N.49TelitaciceptInhibits BAFF and APRILPhase 2 trial clinically meaningful reduction in proteinuria in IgAN. Awaiting phase 3 trial results50 and pediatric study is underway.51PovetaciceptInhibits BAFF and APRILAwaiting phase 1 / 2 trial results in IgAN, membranous nephropathy, lupus nephritis and antineutrophil cytoplasmic autoantibody (ANCA)-associated vasculitis.52 RAINER phase 3 trial in IgAN awaiting completion.BortezomibDepletes plasma B cellsCase studies in IgAV and IgAN53^,^54DaratumumabTargets CD38Trials conducted in multiple glomerular diseases with early promising results.55Targeting complement pathwaysAvacopanComplement 5a receptor inhibitorCompleted pilot phase 2 in IgAN56 and successful trial in ANCA vasculitis.57PegcetacoplanComplement 3 factor inhibitorPhase 2 trial in C3 glomerulopathy and phase 3 findings awaited58IptacopanComplement factor B inhibitorReduction in proteinuria seen in the phase 3APPLAUSE-IgAN trial59 and phase 3 trials in C3 glomerulopathy have successfully met end points.60^,^61CemdisiranComplement 5 factor inhibitorCompleted phase 2 trial in IgAN with reduction in proteinuria.62RavulizumabTerminal pathway activation, complement 5 factor inhibitorPhase 2 clinical SANCTUARY Study demonstrates early sustained improvements in proteinuria in IgAN.63 ICAN Phase 3 trial awaiting completion includes patients with IgAN and inactive IgAV-N. Pediatric IgAN study underway.64Sefaxersen (IONIS -FB-LRx)Antisense oligonucleotide inhibitor of complement factor B.Early phase trials positive with progression to phase 2 and 3 (IMAGINATION) trials underway for IgAN.65NarsoplimabMASP-2 inhibitorARTERMIS-IgAN phase 3 trial terminated at interim analysis due to lack of efficacy in IgAN.Targeting other pathwaysSparsentanDual endothelin and angiotensin receptor antagonistPROTECT trial in IgAN demonstrated efficacy,66 phase 2 pediatric study on glomerular proteinuria (EPPIK) underway and includes patients with IgAV.AtrasentanSelective endothelin A receptor inhibitorALIGN trial in IgAN is underway67TocilizumabIL-6 receptor blockerCase report in IgAV,68 used in other types of rheumatological conditions.Targeted release budesonideGlucocorticoid that specifically targets the gut associated lymphoid tissue of the small intestine.Trials in IgAN show reduction of proteinuria as add-on to standard of care.69 Formulation not suitable for pediatric GI tract and uncertain origin of Gd-IgA in IgAV.Gd-IgA; galactose-deficient-IgA1; IgAN, IgA nephropathy; IgAV, IgA vasculitis; IgAV-N, IgAV associated with nephritis.
Conclusion
Despite major recent advances in other forms of glomerulonephritis, particularly IgAN, IgAV-N is a highly neglected rare disease in terms of evidence-based therapeutic options. This manuscript reports the proceedings of a multiprofessional, collaborative workshop of international experts to develop collective solutions toward generating high-quality evidence for patients with IgAV-N. The global attendees each brought a unique perspective and a shared vision of creating a pipeline of age-inclusive, high-quality clinical trials starting with feasible phase 2 studies to facilitate progression into confirmatory phase 3 studies. The key action points generated from the meeting are outlined in Table 2; and through collaborative efforts, they are being completed to achieve this ambition. Overall, there was an overwhelming desire to change the paradigm for this condition, and this manuscript serves as a benchmark toward establishing a comprehensive path to generating high-quality evidence for patients with IgAV-N.Table 2A summary of the key priorities to support progression toward high quality clinical trials for IgAV-NKey componentsActionsEnhancing data and aligning clinical careObservational cohort dataTo enrich the published literature using established registries from multiple sources.Disease specific scoring tool to objectively phenotype patients.To define and undertake the methodology to improve and achieve validation of a disease specific vasculitis activity scoring tool for IgAV.Define the cost of managing patients with IgAV.Develop the methodology to conduct a formal health economic analysis of this condition.Align clinical carePromote the use of internationally agreed clinical practice guidelines.Improving the scientific understandingDemonstrate biological similarity with IgAN.Use multiomic techniques to evaluate the granular details between the diseases, for example spatial transcriptomics and proteomics of the kidney biopsy tissue.Understand and compare the sites of production of galactose-deficient IgA1 (Gd-IgA1) in IgAN and IgAV in adults and children.Conduct mechanistic studies to determine the origin of Gd-IgA1 and its connection with potential triggers such as epipharynx-kidney axis.Evaluate whether Gd-IgA1 or other markers such as complement pathway products relate to disease activityEvaluate complement deposition in biopsy tissue and complement components in urine and correlate these with histologic (e.g., Oxford M, E, C scores) and clinical (e.g., proteinuria, hematuria, eGFR slope) markers of disease activity and progression.Pathological classificationWork with international networks (e.g., Renal Pathology Society, International IgA Nephropathy Network Research Group, IPNA, KDIGO) to establish a formal statement on the best practice in reporting and classifying the kidney histological features in this disease.Progression to clinical trialsStrengthen industry partnershipsInvest in events related to this condition to develop a positive research environment for collaboration.Suitable agentsCreate criteria to select potential agents to evaluate in this condition.Securing funding for a clinical trialExperts to design trial protocols and seek country specific funding opportunities using a mirrored approach to avoid duplication.Regulatory approved end points and trial designTo design a draft trial protocol using expert input and establish international regulatory approval particularly for the end point selection.PeoplePatient engagementTo work with patient associations to highlight the lack of progress in improving this disease, the poor outcomes reported, and adult-onset disease to enhance the need for evidence. Encourage country specific patient association groups to unite internationally.Determine the most appropriate patient-centered outcomes and end pointsIn partnership with patients and parents/caregivers to elicit the most important symptoms and functional impacts of IgAV in a life course approach that may be addressed with treatment in clinical research and use the results to identify or develop appropriate patient- or observer-reported outcome measures.Engaged expert communityInvite attendees to form an international clinical trial working group for the acceleration of IgAV evidence.eGFR, estimated glomerular filtration rate; Gd-IgA1, galactose-deficient-IgA1; IgAV, IgA vasculitis; IgAV-N, IgAV associated with nephritis; KDIGO, Kidney Disease: Improving Global Outcomes.
Disclosure
LO provides expert consultancy for Aurinia Pharmaceuticals, Eloxx, Dimerix, Biohaven Pharmaceuticals Inc, Santhera Pharmaceuticals, Hansa biopharm, Boehringer-Ingelheim, Sandoz, Biocryst pharmaceuticals Ltd, Novartis, Bedrock, Chinook therapeutics, Roche products Ltd, Travere therapeutics Ltd, and Alentis therapeutics. SRB has received honoraria, Speakers Bureau, and Educational Grants from Vifor and Otsuka. CS has received honoraria and advisory board work for GSK and Vifor. MH has received consulting fees from Novartis, AstraZeneca, Argenx, Otsuka, Biogen, Travere, and Vera Therapeutics; and has received research funding from Argenx. LH has received honorarium from GSK, CSL Vifor, and AZ; and research funding from Vifor and MSD. WYD has equity in Purespring Therapeutics. DJ has received consultancy fees and honoraria from Alentis, Amgen, AstraZeneca, Aurinia, Boehringer, GSK, Novartis, Otsuka, Roche/Genentech, Takeda, and CSL Vifor; and research grants from GSK, Roche/Genentech, and CSL Vifor. JW has received PhD funding via the Cambridge-GSK Translational Immunology Collaboration (CG-TIC). CKC reports consulting or speaker fees from Alexion, Boehringer Ingelheim, Calliditas, CSL Vifor, Dimerix, Emerald Clinical, Novartis, Otsuka Pharmaceutical, Roche, Sobi, Stada, Travere Therapeutics, Vera Therapeutics, and Vertex; and research funding from Travere Therapeutics. ZA works for Vasculitis UK and has received honorarium and small payments for contributing in committees or reviewing materials from CSL Vifor and NOVARTIS. All the other authors declared no competing interests.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1He G.Tao L.Li C.Zhong X.Wang H.Ding J.The spectrum and changes of biopsy-proven kidney diseases in Chinese children J Nephrol 36202341742710.1007/s 40620-022-01527-236472788 · doi ↗ · pubmed ↗
- 2Pillebout E.Sunderkötter C.Ig A vasculitis Semin Immunopathol 43202172973810.1007/s 00281-021-00874-934170395 · doi ↗ · pubmed ↗
- 3Ozen S.Pistorio A.Iusan S.M.EULAR/PRINTO/PRES criteria for Henoch-Schönlein purpura, childhood polyarteritis nodosa, childhood Wegener granulomatosis and childhood Takayasu arteritis: Ankara 2008. Part II: Final classification criteria Ann Rheum Dis 69201079880610.1136/ard.2009.11665720413568 · doi ↗ · pubmed ↗
- 4Gage A.Pepper R.J.Marro J.Salama A.D.Oni L.Ig A vasculitis across the ages: is it time for a precision medicine approach?ACR Open Rheumatol 72025 e 7008310.1002/acr 2.70083 PMC 1242676540937635 · doi ↗ · pubmed ↗
- 5Koskela M.NihtiläJ.Ylinen E.HLA-DQ and HLA-DRB 1 alleles associated with Henoch-Schönlein purpura nephritis in Finnish pediatric population: a genome-wide association study Pediatr Nephrol 3620212311231810.1007/s 00467-021-04955-733591409 PMC 8260528 · doi ↗ · pubmed ↗
- 6Heineke M.H.Ballering A.V.Jamin A.Ben Mkaddem S.Monteiro R.C.Van Egmond M.New insights in the pathogenesis of immunoglobulin A vasculitis (Henoch-Schönlein purpura)Autoimmun Rev 1620171246125310.1016/j.autrev.2017.10.00929037908 · doi ↗ · pubmed ↗
- 7Nüsken E.Weber L.T.Ig A vasculitis nephritis Curr Opin Pediatr 34202220921610.1097/MOP.000000000000112035125382 · doi ↗ · pubmed ↗
- 8Liao C.H.Tsai M.Yang Y.H.Chiang B.L.Wang L.C.Onset age is a risk factor for refractory pediatric Ig A vasculitis: a retrospective cohort study Pediatr Rheumatol Online J 1820208610.1186/s 12969-020-00480-333172497 PMC 7654143 · doi ↗ · pubmed ↗