TAFRO syndrome requiring combined IL 6 and IL 1 inhibition: a case report
Ahmad Wael Sultan, Eva Grießhammer, Carl Hinrichs, Lars Trenkmann, Ilske Oschlies, Leif Gunnar Hanitsch, Hildrun Haibel, Thomas Schneider, Rasmus Leistner

TL;DR
A young man with a rare and severe immune disease called TAFRO syndrome needed a combination of two anti-inflammatory drugs to achieve long-term remission.
Contribution
This case report demonstrates the necessity of combined IL-6 and IL-1 inhibition for treating refractory TAFRO syndrome.
Findings
The patient showed complete remission with combined IL-6 and IL-1 inhibition.
Monotherapy with IL-6 inhibition alone led to disease flare-ups.
The case highlights the diagnostic challenges of TAFRO syndrome due to its non-specific symptoms.
Abstract
TAFRO syndrome is a rare and severe variant of idiopathic multicentric Castleman disease (iMCD). The name-giving presentation is a combination of thrombocytopenia, anasarca, fever, reticulin fibrosis/renal dysfunction, and organomegaly. The disease’s complex but unspecific presentation shows overlapping features with hyperinflammation syndromes of infectious, malignant, or autoimmune origin. Here, we present a case of a young patient with iMCD TAFRO refractory to targeted IL-6 inhibition. A 21-year-old previously healthy male developed progressive systemic inflammation with fever, persistent generalized lymphadenopathy, bicytopenia, polyserositis, and renal failure. Infectious diseases and oncological and immunological workup within the first 6 months after symptom onset yielded an unclear hyperinflammatory syndrome. Subsequently, the patient was treated with IL-1 inhibition resulting…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
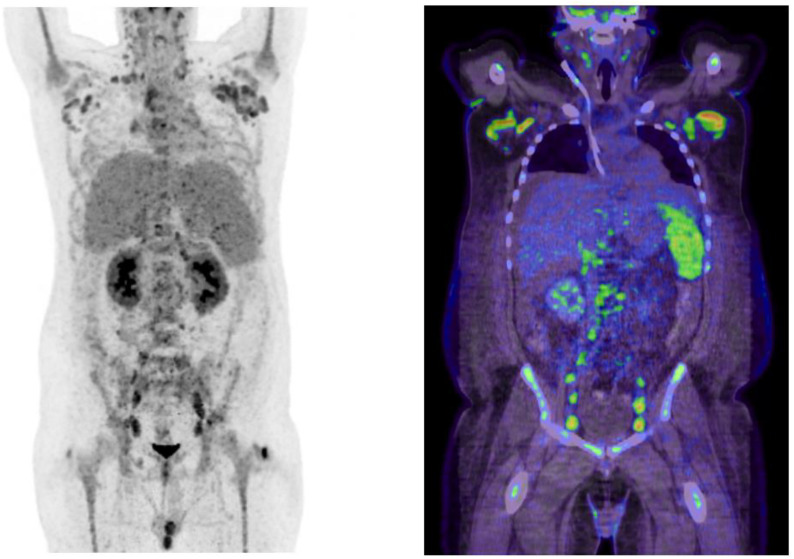 Figure 1
Figure 1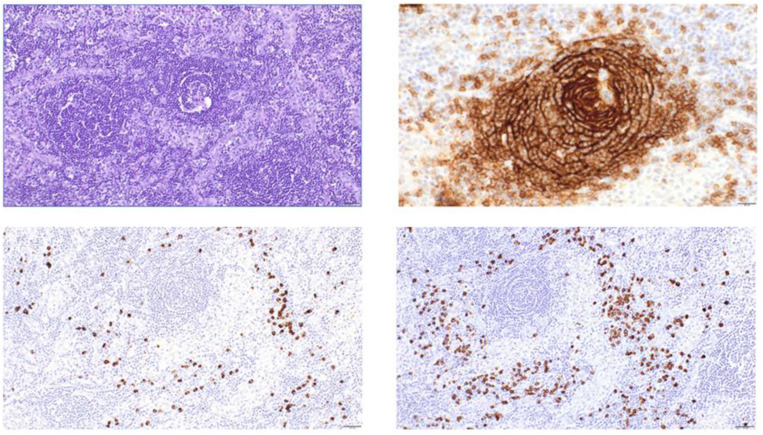 Figure 2
Figure 2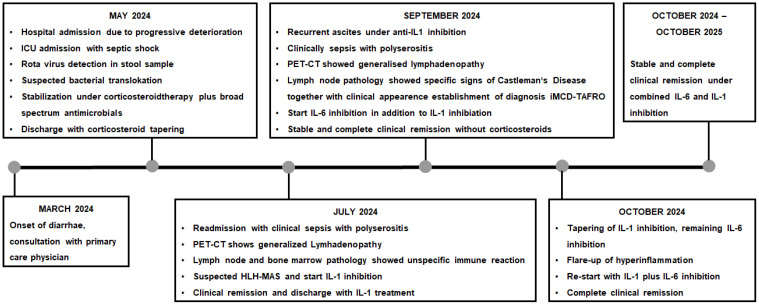 Figure 3
Figure 3| Laboratory parameter | Result | Normal range |
|---|---|---|
| Leukocytes | 25.7 × 10³/μL | 3.7 – 9.2 × 10³/μL |
| Hemoglobin | 7.5 g/dL | 12.0 – 17.9 g/dL |
| Thrombocytes | 27 × 10³/μL | 154 – 401 × 10³/μL |
| CRP | 335/mg/dL | <0.5mg/dL |
| eGFR | 22 mL/min | >90 mL/min |
| Albumin | 18 g/L | 35-52 g/L |
| ferritin | 3,571 ng/mL | 30–400 ng/mL |
| sIL-2R | 16,700 IU/mL | 223–710 IU/mL |
| IL-6 | 124 pg/mL | <7 pg/mL |
| VEGFa | 998 pg/ml | 30–883 pg/mL |
| Feature | iMCD-TAFRO | HLH | Other mimics (iMCD-NOS, autoimmune, lymphoma) |
|---|---|---|---|
| Clinical presentation | Acute onset; fever; anasarca; abdominal pain; organomegaly (hepatosplenomegaly); may lack prominent lymphadenopathy | Acute onset; fever; hepatosplenomegaly; cytopenias; neurologic symptoms; rash | Subacute/chronic; variable fever; lymphadenopathy; organomegaly; less anasarca |
| Thrombocytopenia | Marked, often transfusion-resistant | Marked, part of bicytopenia | Mild or absent; iMCD-NOS may have thrombocytosis |
| Anasarca/edema | Prominent, often with pleural effusions | May occur, but less prominent | Rare; may occur in severe autoimmune disease |
| Fever | High, persistent | High, persistent | Variable; less common in indolent mimics |
| Organomegaly | Hepatosplenomegaly, mild lymphadenopathy | Hepatosplenomegaly, lymphadenopathy | Lymphadenopathy prominent in lymphoma/iMCD-NOS |
| Renal dysfunction | Common, often with proteinuria | May occur (secondary to multiorgan failure) | Variable; less common |
| Hemophagocytosis | Absent or rare | Characteristic (bone marrow, other tissues) | Absent |
| Hyperferritinemia | Mild to moderate | Marked, often >10,000 ng/mL | Mild or absent |
| CRP/ESR | Elevated CRP, variable ESR | Elevated CRP, variable ESR | Elevated in inflammation |
| IgG/hypergamma-globulinemia | Normal or low IgG; polyclonal gammopathy rare | Normal or low IgG | iMCD-NOS: polyclonal hypergammaglobulinemia |
| Anti-SSA/Ro antibodies | Often positive | Negative | Negative |
| Imaging | Mild lymphadenopathy; marked effusions/anasarca; organomegaly | Hepatosplenomegaly; lymphadenopathy; CNS lesions (MRI) | Prominent lymphadenopathy (lymphoma); variable effusions |
| Bone marrow | Reticulin fibrosis; mild plasmacytosis | Hemophagocytosis; cytopenias | Plasmacytosis (iMCD-NOS); malignant cells (lymphoma) |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsViral-associated cancers and disorders · Kawasaki Disease and Coronary Complications · IgG4-Related and Inflammatory Diseases
Introduction
TAFRO syndrome is considered a fulminant, hyperinflammatory manifestation of idiopathic multicentric Castleman’s disease (iMCD TAFRO). The pathognomonic combination of thrombocytopenia, anasarca, fever, reticulin myelofibrosis (or renal insufficiency), and organomegaly have given the disease its name (1–3). The diagnosis is based on clinical signs, symptoms, and specific histopathological findings (3–9). The 5-year survival rate for iMCD TAFRO is approximately 66%, with most deaths occurring within the first few months after diagnosis (3, 5, 10). However, based on the complexity of the diagnosis, the likelihood of underreported mortality is high (1, 2). Although IL-6 inhibition provides a relevant treatment option, the likelihood of sustained remission with this treatment can be achieved in only 30%-50% of patients. Most patients require ongoing therapy to maintain disease control (1, 3, 5).
iMCD TAFRO typically presents with fulminant sepsis-like features, generalized lymphadenopathy and a rapid deterioration (5, 11). Initially, patients are usually treated in the intensive care unit. They are initially treated for fulminant bacterial infections, but if their condition worsens, they often receive immunomodulatory treatment (5, 10, 12, 13). To approach the diagnosis, infections, autoimmune diseases and malignancies with similar clinical presentation must be evaluated. In the light of the diagnosis Castleman disease the most common pathogenetic driver, HHV-8 infection in HIV-positive patients, must be excluded. Further important differential diagnosis in HHV-8 negative cases are cytokine storm syndromes such as hemophagocytic lymphohistiocytosis (HLH) (3). In contrast to iMCD, HLH is associated with significantly higher levels of ferritin and pronounced hemophagocytosis in lymph node and/or bone marrow (3, 4, 9, 14).
In iMCD TAFRO, IL-6 acts as a central proinflammatory mediator. It drives the activation of the PI3K/AKT/mTOR signaling pathway, which correlates with disease severity, capillary leakage, and anasarca and therefore acts as the main therapeutical target (15). However, in approximately 50% of cases, IL-6 inhibition alone does not provide sufficient and sustained treatment success (3, 5, 13). Additional IL-1 inhibition—although justified on pathophysiological grounds and recommended by few experts (3)—has so far only been documented in individual cases (16).
Patient information and clinical findings
A previously healthy 21-year-old Caucasian man developed in April 2024 diarrhea and abdominal pain. As symptoms progressed, including fever >39 °C, increasing fatigue, and persistent gastrointestinal complaints, he was admitted to a regional hospital. During the initial workup, stool testing confirmed rotavirus infection. Within days, he developed the picture of septic syndrome, which led to the suspicion of bacterial translocation, secondary to viral gastroenteritis. Despite thorough microbiological workup, no relevant pathogen was found. The patient remained febrile, and inflammatory markers continued to rise despite broad-spectrum antimicrobial therapy. Moreover, hemodynamic status and renal function progressively worsened, necessitating vasopressor support and hemodialysis. A CT scan showed generalized lymphadenopathy, splenomegaly, and polyserositis including pleural effusion and ascites. Due to his continuing deterioration despite intensive therapeutic efforts, he was transferred to a tertiary care center.
As the clinical course worsened further, the patient was started on immunosuppressive therapy including pulsed corticosteroids (1 mg/kg prednisolone equivalent/day) and immunoglobulin. The overall presentation then raised suspicion of an underlying hyperinflammatory syndrome, notably hemophagocytic lymphohistiocytosis (HLH) with an H-score of 146 and an overall moderate probability for this diagnosis (14, 17). Based on the suspected HLH, a treatment with the IL-1 receptor antagonist anakinra was initiated (200mg s.c./day). Under this regimen, the patient’s condition improved the corticosteroids could be tapered, allowing step-down to non-intensive care and eventually transfer to rehabilitation in August 2024.
Despite initial stabilization, the patient suffered another flare of systemic inflammation 1 month later. This time, the leading symptom was massive ascites necessitating drainage of 16-L sterile ascites within 10days. A PET CT confirmed hypermetabolic lymphadenopathy and splenomegaly (Figure 1). An excision of an axillary lymph node was performed and submitted to the National Reference Center for Lymphoma Studies. This time, histopathological analyses revealed pathognomonic signs of leading to the diagnosis iMCD TAFRO (Figure 2). Figure 3 illustrates the timeline of the clinical course of disease, including key diagnostic and therapeutic milestones (Figure 3).
PET CT imaging revealed generalized hypermetabolic lymphadenopathy involving cervical, mediastinal, axillary, abdominal, and inguinal lymph nodes, consistent with multicentric Castleman disease.
Histopathology of the lymph node revealed regressive changes of the B-cell follicles and an increase in interfollicular small vessels (A, periodic acid-Schiff-staining (PAS) ×200). The regressive B-cell follicles lack germinal centers, and onion-shaped proliferations of the processes of follicular dendritic cells can be observed within the follicle mantle (B, CD21 × 400). An interfollicular increase in polytypic plasma cells is present (C, lambda- ×200 and D, kappa- × 200, in-situ hybridization).
Course of disease from onset of symptoms until complete remission.
Timeline
Diagnostic assessment
Laboratory testing showed leukocytosis (25.7 × 10³/μL), anemia (hemoglobin 7.5 g/dL), thrombocytopenia (27 × 10³/μL), CRP 335 mg/dL, impaired renal function (eGFR 22 mL/min), hypoalbuminemia (17.7 g/L), elevated ferritin (3,571 ng/mL), soluble interleukin-2 receptor (sIL-2R, 16,700 IU/mL), interleukin-6 (IL-6, 124pg/mL), and vascular endothelial growth factor (VEGFa, 998pg/mL) but no relevant gammaglobulinemia (Table 1). Infectious disease workup, among others HIV, HHV-8, EBV, CMV, toxoplasmosis, tbc, and parvovirus B19, yielded negative results. The workup for autoantibody diseases, notably ANA, anti-ENAs, ANCAs, IgG4, and anti-ds-DNA, showed a negative result. Due to sustained renal dysfunction, transvenous kidney biopsy was performed, demonstrating secondary thrombotic microangiopathy (TMA), compatible with renal involvement in the hyperinflammatory process. A positron emission computed tomography (PET CT) confirmed metabolically active lymphadenopathy in supra- and infradiaphragmatic regions, along with hypermetabolic splenomegaly (Figure 1). The patient showed persistent bicytopenia (erythrocytes and thrombocytes); this prompted a bone marrow puncture and lymph node biopsy. The results initially showed signs consistent with reactive inflammation without further specific results.
Only the repeated lymph node histopathology then revealed grade 2 regressive follicles and reactive plasma cell infiltration (Figure 2), fulfilling one major diagnostic criterion for multicentric Castleman disease (MCD). Immunohistochemistry demonstrated preserved follicular dendritic cell networks with few plasma cells expressing IgG and HHV-8 negativity. This time, based on the histopathological results, together with the clinical presentation, the diagnosis of idiopathic multicentric Castleman disease with TAFRO Syndrome (iMCD TAFRO) was established. The diagnosis was based on the fulfillment of 2017 consensus criteria (major criteria: histopathologic lymph node features consistent with the iMCD spectrum plus generalized lymphadenopathy; minor criteria: elevated CRP, anemia, thrombocytopenia, hypoalbuminemia, renal dysfunction, polyclonal hypergammaglobulinemia, B-symptoms, splenomegaly, and polyserositis; exclusion criteria: HHV-8/EBV/CMV/HIV/toxoplasmosis/tbc negativity, exclusion of rheumatological disorder based on overall negative screening for autoantibodies, exclusion of malignant disorders by repeated histopathological examination of enlarged lymph nodes and bone marrow, notably without a sign of hematophagocytosis) (8). Based on the proposed severity score by Masaki et al., the patient reached a score of 11 of 12 being representative for grade 5 (highest severity) (6, 18). Genetic testing included whole exome sequencing (WES) using next-generation sequencing (NGS) and revealed a heterozygous variant of unknown significance in PI3KCD (NC_000001.11:g.9720854C>T; NM_005026.5:c.1634C>T; p.Ala545Val).
Therapeutic intervention, outcome, and follow-up
Based on the diagnosis of iMCD TAFRO, IL-6 inhibition with tocilizumab (8mg/kg i.v. every 4 weeks) in addition to the established IL-1 blockade (100mg s.c./day) was started. We chose tocilizumab over siltuximab as this agent is routinely applied in our department, which was quickly available and could safely be administered (19). Under this combined therapy, the patient showed rapid and substantial clinical improvement and remained clinically stable with normalizing laboratory values and no clinical signs of disease recurrence. In order to achieve the minimum therapy necessary, we attempted to gradually taper anakinra. Unfortunately, this led to another inflammatory flare-up. After anakinra was reinitiated with the dose of 100mg s.c./day, we observed again rapid recompensation. The patient is still under close and frequent outpatient surveillance without further antimicrobial prophylaxis. However, combined immunotherapy has not shown signs of infection which would justify a general antimicrobial prophylaxis. After more than 12 months of follow-up, he still remains stable and at home.
Discussion
This case illustrates diagnostic and therapeutic challenges of idiopathic multicentric Castleman disease with TAFRO syndrome (iMCD TAFRO). Our patient exhibited the characteristics of acute life-threatening systemic inflammation with pronounced ascites and generalized lymphadenopathy.
The clinical presentation and initial investigations pointed to non-specific lymphomatous diseases but also to possible cytokine storm syndromes such as hematophagocytic lymphohistiocytosis (HLH). However, the differential diagnosis of HLH remained a priori unlikely. Only significantly higher ferritin levels (>10,000 ng/mL) provide a sufficiently specific marker for HLH. Furthermore, histopathological examinations revealed no signs of hemophagocytosis (17). However, we found signs that are representative of the hyaline vascular subtype of Castleman disease (HV). Table 2 provides a concise comparison of signs and symptoms seen iMCD TAFRO, HLH, and other most relevant differential diagnoses.
The important distinction between secondary Castleman disease and idiopathic variants was made by immunohistochemical exclusion of HHV-8 infection (4, 8, 9). Within the entity of iMCD, there are three relevant forms: iMCD TAFRO, iMCD with plasmacytic lymphadenopathy (iMCD IPL), and iMCD not otherwise specified (NOS) (9, 20). The clinical presentation specifically met the prerequisites of iMCD TAFRO (3, 9). In iMCD TAFRO, the lymphohistopathological types HV and mixed (hyaline vascular and plasmatic cells, HV/PC) have been predominantly observed. In contrast to that, iMCD IPL cases appear to be primarily plasma cell (PC) driven, whereas iMCD NOS apparently is a collection of heterogeneous cases, clinically distinct from TAFRO and histopathologically distinct from iMCD IPL (20, 21).
The pathogenesis of iMCD involves a complex interplay of dysregulated cytokine signaling with IL-6 as a central mediator. Whereas in iMCD TAFRO IL-6 is mainly expressed by endothelial cells, in iMCD IPL, plasma cells are the main IL-6 drivers (21). The IL-6 overproduction activates downstream signaling pathways such as PI3K/AKT/mTOR and other immune cells (2, 3). In iMCD TAFRO, a pronounced activation of the PI3K-Akt signaling pathway is associated with increased angiogenesis and vascular permeability (22). Interestingly, post-hoc genetic testing of our patient identified a heterozygous variant of unknown significance (VUS) in the phosphatidylinositol‐4,5‐bisphosphate 3‐kinase, catalytic subunit delta gene (PI3KD) (23). Phosphoinositide 3-kinases, particularly the PI3KD pathway, play a crucial role in regulating immune functions affecting, e.g., T- and B-cell function (24). The found VUS in PIK3CD is rare and has not been previously published. Functional data for this variant is not known. Pathogenic variants in PIK3CD are, e.g., associated with activated PI3Kdelta syndrome (APDS) (25). This is a gain-of-function disorder leading to constitutive activation of this specific pathway and downstream hyperactivation of AKT and mTOR signaling. This results in immune dysregulation, lymphoproliferation, and increased risk of lymphoma, as well as recurrent infections and autoimmunity. In IL-6 refractory iMCD, mTOR activation is a recognized molecular signature, with increased phosphorylation of mTOR effectors in affected lymph nodes and peripheral immune cells (15). This hyperactivation is mechanistically plausible given the convergence of cytokine signaling. Thus, mTOR inhibition with sirolimus is an evidence-based option for IL-6-refractory iMCD. Clinical studies demonstrated durable remissions and biochemical improvement in patients who failed anti-IL-6 therapy (26).
The main treatment options in iMCD include high-dose glucocorticoids followed by IL-6 inhibition. Siltuximab is currently the only FDA-approved IL-6 blocker for Castleman disease (19, 27, 28). The progression-free survival associated with this treatment was 15 months (29). However, iMCD TAFRO cases show in about 50% insufficient treatment results under IL-6 inhibition, thus requiring second-line options (2, 3, 5, 11). Second-line treatments are cyclosporine A, rituximab, and cyclophosphamide (3, 10). However, there are no randomized, controlled studies to date. The available evidence for TAFRO is based exclusively on retrospective cohorts (12, 13, 30).
Considering therapeutic strategies and outcomes, there seem to be relevant differences in the studied Japanese and Western iMCD-TAFRO cohorts (10, 13). Japanese patients show higher disease activity and mortality. TAFRO is recognized there as a distinct, severe inflammatory syndrome treated with corticosteroids, tocilizumab, cyclosporin A, or rituximab. In Western countries, iMCD-TAFRO is considered a subtype of iMCD, with siltuximab as standard first-line therapy. A recently published study showed the effectivity of ruxolitinib (JAK1/2 inhibitor) in combination with ropeginterferon alfa-2b. This could be a promising new option for refractory TAFRO-like syndrome. However, further studies are needed to define their optimal use and long-term outcomes (31).
In our case, an earlier initiated IL-1 inhibition led to partial disease. Given his critical condition, we continued this treatment but additionally started IL-6 inhibition. This combination therapy led to the complete remission of symptoms. However, in an attempt to reduce IL-1 inhibition and switch to anti-IL-6 monotherapy, our patient developed a flare-up of symptoms. After resuming treatment, the symptoms completely subsided again, underscoring the importance of the established combination of IL-6 and IL-1 inhibition. There are case reports on the use of IL-1 blockade in patients with iMCD TAFRO as second line (16, 32, 33). IL-1 is known to be a key upstream inducer of IL-6 production through intracellular signaling pathways, also including PI3K/AKT/mTOR (34). However, there are no published studies on combination therapy with IL-1 and IL-6 blockade.
Current adult iMCD guidelines do not provide a formal recommendation for dual IL-1 plus IL-6 blockade in the TAFRO subtype. The use of such a combination therapy is not established in consensus statements or clinical practice standards (3, 11). The available clinical evidence consists primarily of case reports and small series. There, anakinra was seen as a second-line or rescue therapy, most often as monotherapy. For instance, there are some pediatric TAFRO cases with successful high-dose intravenous administration, in patients refractory to IL-6 blockade (16). Mechanistically, there is plausibility for dual blockade: IL-1 can upregulate IL-6, and both cytokines converge on the PI3K/AKT/mTOR pathway, which is implicated in the pathogenesis of IL-6-blockade-refractory iMCD TAFRO (15).
Conclusion
This case illustrates the diagnostic and therapeutic challenges of idiopathic multicentric Castleman disease with TAFRO syndrome (iMCD TAFRO). Treatment with IL-1 blockade initially provided partial symptom control, but sustained remission was achieved only after combination therapy including additional IL-6 inhibitors. The patient’s complete remission demonstrates the efficacy of dual IL-6 and IL-1 inhibition in refractory cases of iMCD TAFRO. Further studies are necessary to prove this concept in other treatment refractory cases.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Masaki Y Arita K Sakai T Takai K Aoki S Kawabata H . Castleman disease and TAFRO syndrome. Ann Hematol. (2022) 101:485–90. doi: 10.1007/s 00277-022-04762-6, PMID: 35044513 PMC 8768434 · doi ↗ · pubmed ↗
- 2Caballero JC Conejero N Solan L Diaz de la Pinta FJ Cordoba R Lopez-Garcia A . Unraveling TAFRO syndrome: an in-depth look at the pathophysiology, management, and future perspectives. Biomedicines. (2024) 12:1076. doi: 10.3390/biomedicines 12051076, PMID: 38791038 PMC 11118735 · doi ↗ · pubmed ↗
- 3Chen LYC Zhang L Fajgenbaum DC . Expert perspective: diagnosis and treatment of castleman disease. Arthritis Rheumatol. (2025). doi: 10.1002/art.43269, PMID: 40457814 PMC 12809887 · doi ↗ · pubmed ↗
- 4Rodriguez Merino L Pomares AA Arce JR Montes-Moreno S . From Castleman disease histopathological features to idiopathic multicentric Castleman disease: a multiparametric approach to exclude potential i MCD histopathological mimickers. J Clin Pathol. (2023) 77:318–323. doi: 10.1136/jcp-2022-208696, PMID: 36690434 · doi ↗ · pubmed ↗
- 5Wu X Zhang X Qian S Shi C Li X Feng X . The experience of diagnosis and treatment for TAFRO syndrome. Ann Hematol. (2023) 102:3515–20. doi: 10.1007/s 00277-023-05435-8, PMID: 37713125 · doi ↗ · pubmed ↗
- 6Nishimura Y Fajgenbaum DC Pierson SK Iwaki N Nishikori A Kawano M . Validated international definition of the thrombocytopenia, anasarca, fever, reticulin fibrosis, renal insufficiency, and organomegaly clinical subtype (TAFRO) of idiopathic multicentric Castleman disease. Am J Hematol. (2021) 96:1241–52. doi: 10.1002/ajh.26292, PMID: 34265103 PMC 9642098 · doi ↗ · pubmed ↗
- 7Masaki Y Kawabata H Takai K Tsukamoto N Fujimoto S Ishigaki Y . 2019 Updated diagnostic criteria and disease severity classification for TAFRO syndrome. Int J Hematol. (2020) 111:155–8. doi: 10.1007/s 12185-019-02780-1, PMID: 31782045 · doi ↗ · pubmed ↗
- 8Fajgenbaum DC Uldrick TS Bagg A Frank D Wu D Srkalovic G . International, evidence-based consensus diagnostic criteria for HHV-8–negative/idiopathic multicentric Castleman disease. Blood J Am Soc Hematol. (2017) 129:1646–57. doi: 10.1182/blood-2016-10-746933, PMID: 28087540 PMC 5364342 · doi ↗ · pubmed ↗