Growth hormone deficiency in three siblings homozygous for a rare GH1 haplotype
Ana Cláudia Ribeiro, Omneya Magdy Omar, Ebtesam Abdalla, Manuel Carlos Lemos

TL;DR
Three siblings with growth hormone deficiency were found to share a rare genetic variant in the GH1 gene, highlighting how noncoding DNA changes can cause endocrine disorders.
Contribution
This is the first report linking a homozygous GH1 promoter haplotype to isolated growth hormone deficiency.
Findings
A homozygous GH1 haplotype with nine SNPs was identified in three siblings with GH deficiency.
The haplotype overlaps regulatory elements and is associated with reduced promoter activity.
No pathogenic coding mutations or deletions were found in the affected individuals.
Abstract
Growth Hormone (GH), secreted by the anterior pituitary gland, is a key regulator of postnatal growth. Mutations in the GH1 gene can lead to isolated GH deficiency (IGHD), a rare disorder characterized by growth failure and severe short stature. The aim of this study was to identify the genetic basis of IGHD in three siblings born to consanguineous parents. Three siblings were diagnosed with short stature due to GH deficiency (stimulated GH peak levels between 0.07 and 0.77 µg/L). To identify their genetic cause, whole-exome sequencing (WES), multiplex ligation-dependent probe amplification (MLPA), and targeted GH1 sequencing was performed. A shared homozygous GH1 haplotype comprising nine single nucleotide polymorphisms (SNPs), spanning the promoter, coding, and 3’ flanking regions, was revealed. The parents were heterozygous carriers of this haplotype. This rare SNP combination…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
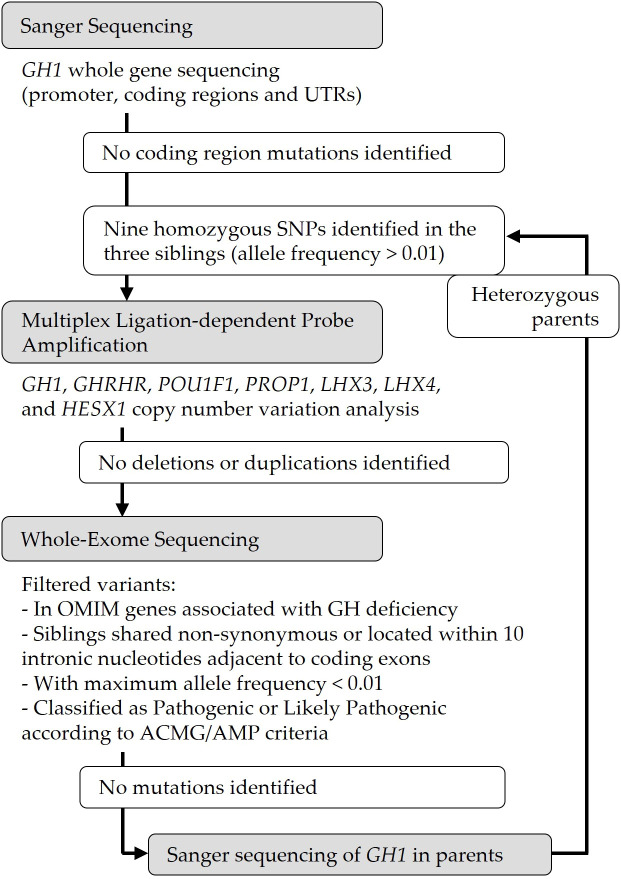 Figure 1
Figure 1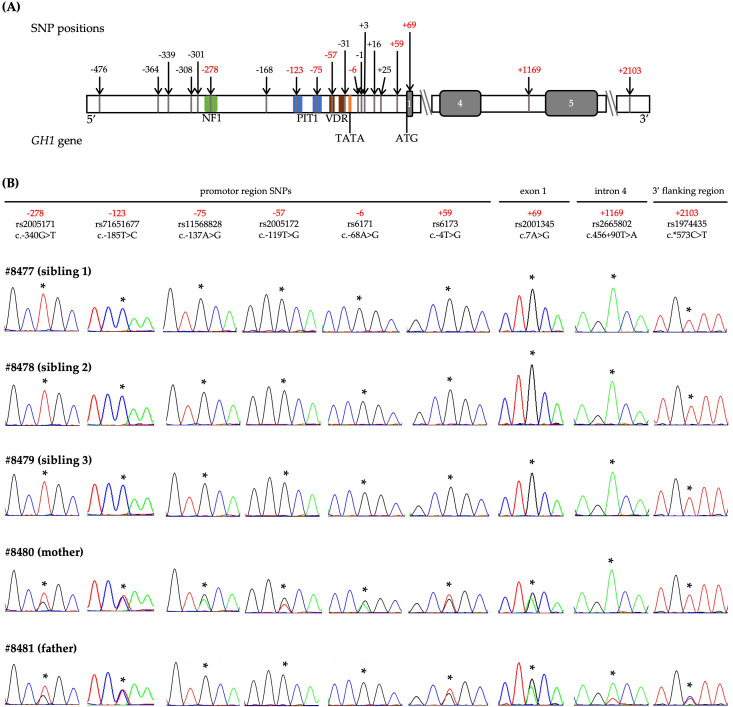 Figure 2
Figure 2| Characteristics | Sibling 1 | Sibling 2 | Sibling 3 |
|---|---|---|---|
| Identification number | #8477 | #8478 | #8479 |
| Gender | Female | Male | Male |
| Current age | 9.6 years | 6.9 years | 4.3 years |
| Age at diagnosis | 5.5 years | 3.5 years | 2 years |
| Bone age (chronological/bone) | 6/4 years | 4/2.5 years | 4/2.5 years |
| GH peak after stimulation (test) | 0.19 µg/L (levodopa) | 0.14 µg/L (levodopa) | 0.77 µg/L (clonidine) |
| Height (Z-score) before treatment | - 4.2 | - 6.4 | - 3.1 |
| Age at start of GH treatment | 6 years | 4 years | Not started yet |
| Initial GH dose | 0.027 mg/kg/day | 0.027 → 0.036 mg/kg/day | |
| Height (Z-score) after treatment | 0.11 (after 43 months) | - 1.4 (after 34 months) | – |
| Brain and pituitary MRI | Normal | Normal | n/a |
| Other clinical problems | None | None | None |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsGrowth Hormone and Insulin-like Growth Factors · Thyroid Disorders and Treatments · Glycogen Storage Diseases and Myoclonus
Introduction
Growth Hormone (GH) plays a fundamental role in human development, growth, and metabolism (1, 2). Its production and secretion by the anterior pituitary gland are highly regulated and complex processes; therefore, any disruption can result in GH deficiency (3). In children, congenital GH deficiency typically presents as decelerated linear growth and is frequently classified as idiopathic (2–4). However, some patients have a genetic cause for their disorder, due to mutations in one of several genes known to cause isolated (IGHD) or combined pituitary hormone deficiency (5, 6).
GH is encoded by the GH1 gene, located on chromosome 17q23.3, within a cluster of five related genes (7). Its expression is regulated by upstream enhancer elements and a promoter region containing multiple transcription factor binding sites (8). The GH1 regulatory and coding regions are highly polymorphic, with over 16 single nucleotide polymorphisms (SNPs) that can form haplotypes influencing transcriptional activity (9, 10). These polymorphisms span the promoter, coding, and noncoding regions, complicating the genetic architecture of GH1 (11).
Mutations in the GH1 coding sequence and whole gene deletions are well-established causes of inherited forms of IGHD, which typically occur without deficiencies of other pituitary hormones (5, 6). Despite extensive studies of GH1 mutations, the cumulative effect of multiple promoter variants has not been linked to monogenic IGHD. Indeed, non-coding regulatory SNPs are often under-investigated, as their precise effect is difficult to determine, requiring extensive functional analysis, which is often not feasible in clinical routine (12). The aim of our study was to determine the genetic cause of IGHD in three siblings born to consanguineous parents.
Material and methods
Patients
We studied five individuals from a consanguineous Egyptian family, including three siblings diagnosed with IGHD and their unaffected parents, who were first cousins. The siblings presented normal motor and cognitive development but exhibited growth retardation and short stature. GH stimulation testing using clonidine and levodopa confirmed GH deficiency in the siblings (Table 1). Other pituitary hormone levels were normal, and brain magnetic resonance imaging (MRI) revealed no structural abnormalities. The two older siblings responded positively to recombinant GH therapy. Clinical data are summarized in Table 1.
The study was conducted in accordance with the Declaration of Helsinki and approved by the Ethics Committee of the Faculty of Health Sciences, University of Beira Interior (Ref: CE-FCS-2012-012). Informed consent was obtained from all subjects involved in this research.
Genetic analysis
Genomic deoxyribonucleic acid (DNA) was extracted from peripheral blood leucocytes using previously described methods (13) and used with polymerase chain reaction (PCR) primers to amplify the GH1 promoter and coding regions (NM_000515.5) (primer sequences and PCR conditions available upon request). Bidirectional sequencing of the PCR products was performed using a semi-automated capillary DNA sequencer (STAB VIDA, Caparica, Portugal; ABI 3730XL, Applied Biosystems; Thermo Fisher Scientific, Waltham, MA, USA). In order to exclude other genetic causes of GH deficiency, we applied additional steps of multiplex ligation-dependent probe amplification (MLPA) and whole-exome sequencing (WES). Specifically, in sibling 2, we analysed copy number variations, through an MLPA assay, using the SALSA MLPA probemix P216-C1 (MRC Holland, Amsterdam, Netherlands), according to the manufacturer’s guidelines. This kit includes probes for GH1, as well as for other GH deficiency-related genes (POU1F1, PROP1, GHRHR, LHX3, LHX4, and HESX1). Fragment analysis was performed with the DNA sequencer described above. In addition, WES was carried out on siblings 1 and 2, as previously described (14). Genetic sequence variants were filtered according to the following cumulative criteria: i) found in both siblings; ii) non-synonymous or located within ten intronic nucleotides adjacent to coding exons; iii) absent or rare (maximum allele frequency < 0.01) in the Genome Aggregation Database (v.4.1.0) (15); and iv) located in genes associated with GH deficiency in the Online Mendelian Inheritance in Man database (16). Variants were classified according to American College of Medical Genetics and Genomics/Association for Molecular Pathology criteria guidelines (17).
Results
Based on the suspicion of autosomal recessive inheritance due to the parental consanguinity, we initially analysed the GH1 gene in the three siblings. No pathogenic mutations were identified in the coding regions or canonical splice sites (Figure 1). However, all three siblings were homozygous for a shared set of nine previously reported SNPs across the GH1 locus: −278T, −123C, −75G, −57G, −6G, +59G, +69G, +1169A, and +2103T, as numbered according to Horan et al. (10) (Figure 2). The population frequency of this haplotype has been estimated to be <1% (10). Both parents were heterozygous for this haplotype. Additional analyses, including MLPA and WES, did not reveal any other potentially pathogenic variants associated with GH deficiency (Figure 1).
Flowchart of the genetic testing of a family with isolated growth hormone deficiency (IGHD). To identify the genetic cause in the siblings, targeted GH1 sequencing, multiplex ligation-dependent probe amplification (MLPA) and whole-exome sequencing (WES) was performed. No pathogenic coding mutations, duplications or deletions were identified. The three siblings were homozygous for a shared set of polymorphisms across the GH1 locus, and both parents were heterozygous for this haplotype. UTRs, untranslated regions; OMIM, Online Mendelian Inheritance in Man; GH, Growth Hormone; ACMG/AMP, American College of Medical Genetics and Genomics and Association for Molecular Pathology.
Genetic analysis of GH1 in a family with isolated growth hormone deficiency. (A) Schematic representation of the GH1 gene showing the positions of 20 known single nucleotide polymorphisms (SNPs) from –476 to +2103, numbered relative to the transcriptional start site, as described by Horan et al., 2003 (10). SNPs identified in the three affected siblings are highlighted in red. Exons are depicted as grey boxes. Coloured boxes indicate transcription factor binding sites: nuclear factor 1(NF1, in green), pituitary transcription factor 1 (PIT1, in blue), vitamin D receptor (VDR, in brown), and the TATA box (orange). (B) Sequence chromatograms of the nine SNP positions for all five family members. Asterisks () indicate the position of each SNP. The three affected siblings were homozygous for the rare haplotype, while both parents were heterozygous, consistent with autosomal recessive inheritance.*
Discussion
Previous studies have demonstrated that polymorphisms in the GH1 promoter can modulate gene expression and influence circulating GH levels (8, 10, 11, 18–23). To date, no homozygous haplotype has been clearly implicated in monogenic IGHD. In this study, we report three siblings with IGHD who share a homozygous GH1 haplotype consisting of nine SNPs, distributed from the promoter to the 3′ flanking region.
Five of the SNPs (−278T, −75G, −57G, −6G, and +59G) comprise the promoter haplotype 21 described by Horan et al. (10). These authors identified 36 different GH1 haplotypes from 15 SNPs (10, 24, 25) in a control population. Haplotype 21 had a population frequency of <1% and demonstrated only 58% promoter activation relative to wild-type in functional assays (10). These SNPs are located in regions essential for GH1 transcriptional regulation (22). The -278T is located within the nuclear factor 1 (NF1) binding site, the -75G in the proximal pituitary transcription factor 1 (PIT1) binding site, the -57G in the vitamin D receptor (VDR) response element region, and the -6G at the transcriptional start site (22). Giordano et al. (19) specifically studied the -75G polymorphism and found reduced GH1 transcription due to decreased PIT1 binding affinity. However, GH stimulation studies in individuals carrying this variant did not confirm a functional effect, likely due to insufficient homozygous sample size (19). These findings support the hypothesis that cumulative effects of multiple regulatory SNPs may reduce GH1 transcription. Indeed, the impaired binding of essential transcription factors, caused by these SNPs, can weaken the promoter’s ability to recruit the necessary machinery for transcription (22).
Our patients also carried four additional SNPs (-123C, +69G, +1169A, and +2103T). The −123C SNP, located in a conserved Specificity Protein 1 (SP1) and PIT1 binding site, has been shown to have no significant impact on transcription in luciferase reporter assays (18, 23). The +69G SNP (p.Thr3Ala) is a missense variant located in the GH signal peptide. Though initially reported in GH-deficient patients (26), it is now considered a benign polymorphism (11, 18, 26, 27). Importantly, the intronic +1169A SNP, in cis with −278T and −57G, has been associated with reduced GH secretion and lower insulin-like growth factor 1 (IGF1) levels (28). Functional studies have demonstrated impaired GH1 expression and GH secretion linked to this variant, although the effect may be influenced by adjacent regulatory SNPs (29). The +2103T SNP, located in the 3′ flanking region, has also been implicated in reduced GH secretion when present with −278T, −57G, and +1169A (29). Notably, Yamamoto et al. (30) identified this four-SNP combination in GH-deficient siblings, although their homozygous mother had normal stature, suggesting these variants alone may be insufficient to cause disease.
In our study, the homozygous co-occurrence of all nine variants — many with individually demonstrated or suspected functional impact — strongly suggests a cumulative regulatory effect on GH1 expression. This may explain the phenotype observed in our patients and supports a role for the genetic screening of GH1 promoter haplotypes in cases of IGHD lacking coding mutations.
Limitations of our study include the absence of in vivo functional data confirming reduced GH secretion associated with this haplotype. Such functional assays would be useful to validate our findings. While MLPA and WES excluded known GH deficiency-associated variants, we cannot rule out the involvement of unknown regulatory elements or genes. Nonetheless, the identification of a shared homozygous rare haplotype in three affected siblings provides compelling evidence for its pathogenicity.
In conclusion, we identified a rare homozygous GH1 haplotype comprising nine regulatory and coding SNPs that may collectively impair GH1 expression, representing the likely cause of IGHD in three siblings from a consanguineous family.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Moller N Jorgensen JO . Effects of growth hormone on glucose, lipid, and protein metabolism in human subjects. Endocr Rev. (2009) 30:152–77. doi: 10.1210/er.2008-0027, PMID: 19240267 · doi ↗ · pubmed ↗
- 2Ranke MB Wit JM . Growth hormone - past, present and future. Nat Rev Endocrinol. (2018) 14:285–300. doi: 10.1038/nrendo.2018.22, PMID: 29546874 · doi ↗ · pubmed ↗
- 3Di Iorgi N Morana G Allegri AE Napoli F Gastaldi R Calcagno A . Classical and non-classical causes of GH deficiency in the paediatric age. Best Pract Res Clin Endocrinol Metab. (2016) 30:705–36. doi: 10.1016/j.beem.2016.11.008, PMID: 27974186 · doi ↗ · pubmed ↗
- 4Alatzoglou KS Webb EA Le Tissier P Dattani MT . Isolated growth hormone deficiency (GHD) in childhood and adolescence: recent advances. Endocr Rev. (2014) 35:376–432. doi: 10.1210/er.2013-1067, PMID: 24450934 · doi ↗ · pubmed ↗
- 5Giordano M . Genetic causes of isolated and combined pituitary hormone deficiency. Best Pract Res Clin Endocrinol Metab. (2016) 30:679–91. doi: 10.1016/j.beem.2016.09.005, PMID: 27974184 · doi ↗ · pubmed ↗
- 6Vasques GA Andrade NLM Correa FA Jorge AAL . Update on new GH-IGF axis genetic defects. Arch Endocrinol Metab. (2019) 63:608–17. doi: 10.20945/2359-3997000000191, PMID: 31939486 PMC 10522240 · doi ↗ · pubmed ↗
- 7Barsh GS Seeburg PH Gelinas RE . The human growth hormone gene family: structure and evolution of the chromosomal locus. Nucleic Acids Res. (1983) 11:3939–58. doi: 10.1093/nar/11.12.3939, PMID: 6306568 PMC 326017 · doi ↗ · pubmed ↗
- 8Birla S Jyotsana PV Sharma A Khadgawat R Garg M Jain V . Genetic characterization of growth hormone 1 gene in patients with isolated growth hormone deficiency. Indian J Endocrinol Metab. (2012) 16:S 310–2. doi: 10.4103/2230-8210.104071, PMID: 23565410 PMC 3603058 · doi ↗ · pubmed ↗