Giant Cell Arteritis With Central Nervous System Vasculitis Presenting As Binocular Diplopia and Ptosis due to Third Cranial Nerve Palsy
João Casanova Pinto, Manuel G. Costa, Beatriz Fernandes, Carlos Ramalheira

TL;DR
A woman in her 60s with giant cell arteritis showed unusual neurological symptoms, including eye issues, and responded well to corticosteroid treatment.
Contribution
This case highlights an uncommon neurological presentation of giant cell arteritis with central nervous system vasculitis.
Findings
The patient exhibited third cranial nerve palsy with ptosis and binocular diplopia, indicating oculomotor nerve involvement.
MRI findings suggested central nervous system vasculitis with T2-FLAIR abnormalities.
Early corticosteroid treatment led to full clinical remission after four months.
Abstract
We report the case of a woman in her 60s with no notable comorbidities presented with a four-week history of bilateral temporal headache, scalp tenderness, jaw claudication, and sporadic fever. She also reported binocular diplopia for the previous 24 hours. Examination revealed tender superficial temporal arteries (TA) and right-sided third cranial nerve palsy with ptosis. Laboratory tests showed elevated inflammation markers. Cranial and cervical computed tomography (CT) and CT angiography (CTA) were unremarkable. She was treated with a single dose of intravenous methylprednisolone, followed by oral prednisolone. Subsequent TA duplex ultrasound demonstrated artery wall thickness, and TA biopsy confirmed chronic inflammation with disruption of the internal elastic lamina, both consistent with giant cell arteritis. Cranial magnetic resonance imaging (MRI) revealed scattered punctate…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
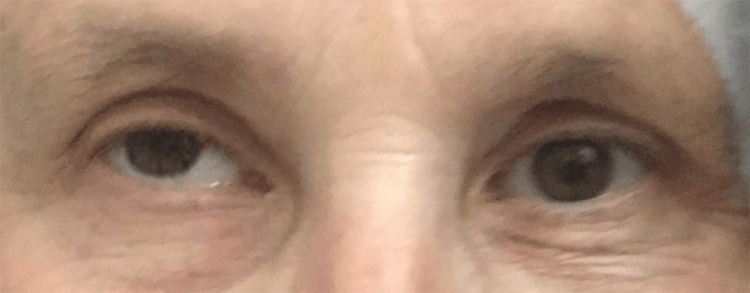 Figure 1
Figure 1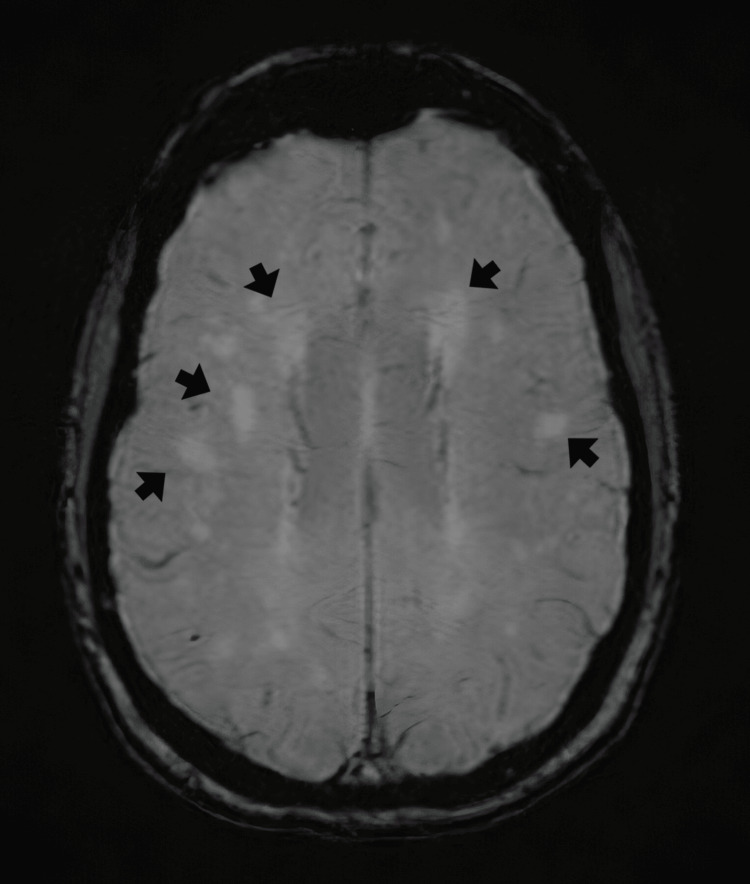 Figure 2
Figure 2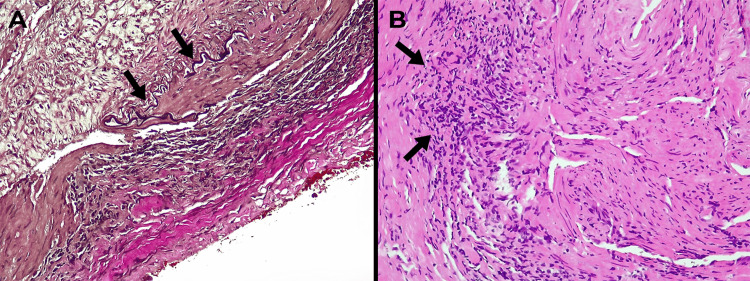 Figure 3
Figure 3| Test | Patient Value | Reference range |
| CRP (mg/dL) | 15.99 | < 0.3 |
| ESR (mm/h) | 48 | < 30 |
| Leucocytes (x 109/L) | 12,500 | 4,500 – 11,000 |
| Neutrophils (x 109/L) | 9,750 | 2,500 – 7,500 |
| Serum creatinine (mg/dL) | 0.81 | 0.6 – 1.1 |
| LDL-cholesterol (mg/dL) | 84 | < 116 |
| Glycosylated hemoglobin (%) | 5.5 | < 5.7 |
| C3 (mg/dL) | 91 | 80 – 180 |
| C4 (mg/dL) | 34 | 15 – 45 |
| Immunological study | Negative for ANA, ANCA, RF, anti-CCP, and antiphospholipid antibodies. | |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsVasculitis and related conditions · IgG4-Related and Inflammatory Diseases · Otitis Media and Relapsing Polychondritis
Introduction
Giant cell arteritis (GCA) is the most common primary vasculitis affecting medium and large arteries in individuals over 50 years of age [1,2]. Typical clinical manifestations include headache, jaw claudication, and visual disturbances, most notably anterior ischemic optic neuropathy (AION). Ocular motor palsies, though less frequent, are well-documented complications arising from ischemia of the vasa nervorum supplying cranial nerves [3]. However, isolated third cranial nerve involvement with ptosis remains a rare and diagnostically challenging presentation, often mimicking benign microvascular cranial nerve palsies or compressive lesions [4].
Concurrent central nervous system (CNS) vasculitis in GCA is an even rarer phenomenon, with only sparse reports linking GCA to inflammatory changes in cerebral vasculature [5]. Recognizing atypical manifestations is key, as delayed treatment may result in severe and irreversible visual complications [2,6]. We report a case of GCA presenting with third cranial nerve palsy, with concurrent imaging findings suggestive of CNS vasculitis. This case highlights the challenges implicated in diagnosing GCA and underscores the importance of timely aggressive corticosteroid therapy.
This case was previously presented as an abstract/poster at the 22nd European Congress of Internal Medicine in Istanbul, Türkiye, 2024.
Case presentation
A woman in her 60s, with no significant past medical history, presented to the emergency department with acute-onset binocular diplopia evolving over 24 hours. Over the preceding four weeks, she had experienced persistent bilateral temporal headache, scalp tenderness, jaw claudication, and intermittent low-grade fever (38.0 ºC). She denied any prior visual disturbances or other neurological deficits, such as facial numbness or hearing changes, as well as any drug intake, weight loss, or constitutional symptoms. There was no history of recent travel, tick bite, or immunosuppression.
On admission, the patient appeared fatigued but alert and oriented. Vital signs were within normal range. Palpation of the superficial temporal arteries revealed marked tenderness and cord-like thickening bilaterally. Neurological evaluation demonstrated partial ptosis of the right eyelid with the right eye deviated outward and upward, resulting in diplopia, consistent with a right oculomotor (third cranial nerve) palsy (Figure 1). Pupillary light reflexes were intact, with no relative afferent pupillary defect. Extraocular movements were otherwise full, and fundoscopic examination did not document disc edema or pallor. The remainder of the cranial nerve examination, motor strength, sensory function, and coordination were unremarkable. The systemic examination, including peripheral pulses, bore no noteworthy findings.
Clinical photograph demonstrating ocular misalignment (strabismus)Image shows right eye outward and upward deviation, consistent with right third cranial nerve palsy.
Initial laboratory studies showed neutrophil-predominant leukocytosis and elevated serum inflammatory markers (Table 1).
Cranial and cervical computed tomography angiography (CTA) did not reveal any aneurysms, stenosis, or occlusive arterial disease. The temporal arteries appeared symmetrically thickened, but no luminal irregularities were noted. Given the clinical suspicion for GCA with ocular involvement, high-dose corticosteroid therapy was initiated with a single dose of 250 mg intravenous methylprednisolone, followed by oral prednisolone at 40 mg daily. Within two weeks, further investigations were performed to confirm the diagnosis and exclude alternative etiologies. The metabolic panel revealed low-density lipoprotein (LDL) cholesterol of 84 mg/dL and a glycated hemoglobin of 5.5%. Blood cultures were negative.
Cerebrospinal fluid (CSF) analysis via lumbar puncture revealed normal opening pressure and absence of cytochemical abnormalities. Cytopathological evaluation, polymerase-chain reaction for Herpesviridae, microbiological cultures, autoimmunity panels, and oligoclonal bands were negative. Serological testing ruled out autoimmune mimics and infectious causes, such as syphilis, hepatitis B, hepatitis C, human immunodeficiency virus (HIV), and tuberculosis.
A thoracic computed tomography (CT) identified a small cluster of old calcified nodules in the right upper lobe, but no evidence of active pulmonary disease. CTA ruled out supra-aortic and aortic abnormalities such as aneurysms or wall thickening. Brain magnetic resonance imaging (MRI) revealed periventricular microangiopathic chronic ischemic changes and scattered hyperintense punctate areas on T2-weighted fluid-attenuated inversion recovery (FLAIR) sequences, bilaterally distributed in the frontoparietal subcortical white matter (Figure 2). These findings were interpreted as microangiopathic changes secondary to vasculitis, with no associated contrast enhancement or mass effect.
Axial T2-weighted FLAIR brain MRI imageBrain MRI revealed several punctate hyperintense areas bilaterally, distributed in the frontoparietal subcortical white matter (black arrows), as well as in a periventricular distribution. These findings were interpreted as microangiopathic changes secondary to vasculitis.FLAIR: fluid-attenuated inversion recovery
Temporal artery duplex ultrasound demonstrated circumferential wall thickening ("halo sign") of the bilateral superficial temporal arteries, with a maximal intima-media thickness of 1.2 mm (reference value: < 0.8 mm). Subsequent temporal artery biopsy, obtained within two weeks of corticosteroid initiation, revealed a chronic inflammatory infiltrate composed predominantly of histiocytes and lymphocytes, with fragmentation of the internal elastic lamina (Figure 3).
Temporal artery biopsy histology(A) Elastic stain (10×) showing marked fragmentation and disruption of the internal elastic lamina (black arrows). (B) Hematoxylin and eosin stain (20×) demonstrating dense transmural lymphohistiocytic inflammatory infiltrate involving the media (black arrows), consistent with active granulomatous arteritis.
Although granulomas and giant cells were absent, these histopathological findings, along with clinical and imaging data, satisfied 16 of the 20 points on the latest European Alliance of Associations for Rheumatology (EULAR) classification criteria for GCA [6]. The patient’s clinical course was notable for rapid improvement: within two weeks of initiating corticosteroids, her headache and jaw claudication completely resolved, and the diplopia significantly improved. Inflammatory markers progressively decreased and ultimately normalized (ESR 1 mm/h, CRP 0.2 mg/dL) by the four-month follow-up, coinciding with a full recovery of right third cranial nerve function.
A structured corticosteroid taper was planned for a total of 52 weeks, considering the one used in the giant cell arteritis Actemra (GiACTA) trial [7]. Prophylactic therapy with alendronate (70 mg/week), vitamin D (2000 IU/day), pantoprazole (40 mg/day), and cotrimoxazole (960 mg thrice/week) was initiated to mitigate glucocorticoid-induced complications. After one year, the patient experienced no relapses of the disease and is currently under prednisolone 5 mg/day. Cranial MRI at the six-month follow-up demonstrated the absence of the initially detected lesions.
Discussion
This case illustrates two rare and clinically significant facets of GCA: third cranial nerve palsy with ptosis and concurrent CNS vasculitis. GCA is a granulomatous vasculitis of medium to large arteries, most often involving branches of the external carotid artery [1,2]. Typical symptoms include headache, mandibular claudication, and visual impairment. Among large-vessel vasculitis, GCA is the leading diagnosis in our patient, supported by her age and classic clinical features [6].
A normal CTA does not exclude GCA, which underscores the importance of temporal artery biopsy as the diagnostic gold standard, despite occasional false negatives, especially if corticosteroid treatment has already commenced [8,9]. Our patient had a positive biopsy despite being under corticosteroids for two weeks. Although visual impairment, typically from AION, remains a well-known complication, ocular cranial nerve palsies may also occur [2,3,9]. While diplopia is present in up to 19% of GCA cases, ptosis from isolated oculomotor nerve involvement is exceptionally uncommon, representing fewer than 2% of reported presentations [8]. Cranial nerve palsies in GCA are thought to result primarily from ischemia of the vasa nervorum or of small arterial branches supplying the nerve, though direct inflammatory involvement of the intracranial vasculature may also play a role [2-4]. In the presence of suspected cranial nerve involvement, brain MRI may show microangiopathic or inflammatory changes in the brain parenchyma, lending further support to the presence of a widespread vasculitis [3].
The confirmation of CNS involvement, although not routine, emphasizes the heterogeneity of GCA presentations [3,10]. In our patient, the frontoparietal T2-FLAIR hyperintense lesions were not confined to the typical periventricular or deep white matter distributions usually associated with chronic hypertensive or age-related small-vessel disease. Instead, the lesions showed a multifocal cortical-subcortical pattern, which, in conjunction with elevated inflammatory markers, cranial neuropathy, and rapid response to immunosuppression, favors an inflammatory vasculitic etiology over isolated chronic microangiopathy [11]. Remarkably, the absence of contrast enhancement or mass effect distinguishes these changes from alternative etiologies such as demyelination, neoplasm, or infectious encephalitis.
The coexistence of GCA and CNS vasculitis remains poorly understood. Both conditions involve inflammation of blood vessels, but they typically affect different vascular beds and have distinct clinical presentations. While GCA primarily involves large and medium-sized cranial arteries, CNS vasculitis affects smaller blood vessels of the brain and spinal cord. However, there are cases where these conditions overlap, suggesting potential shared mechanisms. Both GCA and CNS vasculitis are thought to arise from dysregulated immune responses. In GCA, dendritic cells in the arterial wall activate T-cells (particularly T helper 17 and T helper 1 cells), leading to the production of pro-inflammatory cytokines such as interleukin-6, interleukin-17, and interferon-gamma. These cytokines can promote systemic inflammation, which might extend to the CNS vasculature, triggering or exacerbating CNS vasculitis. The breakdown of the blood-brain barrier (BBB) due to systemic inflammation could allow immune cells and cytokines to infiltrate the CNS, contributing to vasculitis in the brain [12,13].
It has been hypothesized that similar antigens in the vascular walls of cranial and intracerebral arteries could be expressed and lead to cross-reactive immune responses. Molecular mimicry, where pathogens (e.g., viruses or bacteria) express antigens similar to those in human vascular tissue, could also play a role in initiating or perpetuating inflammation in both GCA and CNS vasculitis [14]. Endothelial dysfunction is a common feature in both GCA and CNS vasculitis. In GCA, inflammation leads to intimal hyperplasia and vessel occlusion, while in CNS vasculitis, endothelial injury can result in thrombosis, ischemia, or hemorrhage. Shared mechanisms of endothelial activation, such as upregulation of adhesion molecules and increased expression of pro-inflammatory cytokines, might contribute to the development of vasculitis in both vascular territories [15,16]. Genetic factors might predispose individuals to develop both GCA and CNS vasculitis. For example, human leukocyte antigen DRB1*04 and other immune-related genes may play a role in both GCA and CNS vasculitis, increasing its susceptibility. Familial cases of vasculitis and overlapping genetic risk factors for autoimmune diseases suggest a potential shared genetic basis [17].
Concerning the findings of concurrent CNS vasculitis, a broad range of potential etiologies was considered in the differential diagnosis, including infectious agents, systemic inflammatory/autoimmune diseases, drug-induced processes, neoplastic causes, and other miscellaneous conditions. Infections can trigger a vasculitic process either through direct invasion of the vessel wall or via immune-mediated mechanisms [5]. The patient lacked systemic signs of infection, and blood cultures and serologic testing were negative. Fungal and parasitic infections (e.g., aspergillosis, mucormycosis, neurocysticercosis, malaria) were excluded based on the patient’s immunocompetent status, lack of epidemiologic risk factors, and unsupportive imaging and laboratory results. The patient’s lack of systemic involvement, such as renal impairment, respiratory, joint, or mucocutaneous involvement make the diagnosis of other systemic inflammatory/autoimmune diseases unlikely. Similarly, negative autoantibodies, normal complement values, and absence of other specific radiological findings support GCA-associated vasculitis. In this patient, there was no history of exposure to medications or illicit substances, rendering a drug-induced etiology unlikely.
Paraneoplastic vasculitis associated with hematologic malignancies or solid tumors can occasionally present with vasculitic features [5]. However, a comprehensive evaluation provided no evidence of an underlying neoplastic process in this patient. Additional considerations include reversible cerebral vasoconstriction syndrome (RCVS) and Moyamoya disease. RCVS typically manifests as non-inflammatory vasoconstriction, which is inconsistent with the inflammatory changes observed. Moyamoya disease is characterized by chronic progressive occlusive changes and was not compatible with the acute clinical presentation and imaging findings. Collectively, the synchronized onset of systemic inflammation, cranial neuropathy, and MRI abnormalities strongly supports a unifying diagnosis of GCA with associated CNS involvement, although the absence of histopathological confirmation of cerebral vasculitis (e.g., via brain biopsy) limits definitive attribution.
When dealing with GCA, early recognition and timely administration of high-dose corticosteroids are paramount to prevent permanent visual loss, stroke, or other complications [2,18]. In this case, early initiation of high-dose intravenous methylprednisolone followed by a structured oral prednisolone taper, in accordance with recommendations for high-risk GCA, resulted in rapid clinical improvement, with complete resolution of headache and jaw claudication and marked improvement of diplopia within two weeks of treatment initiation. Inflammatory markers progressively declined and fully normalized by the four-month follow-up (ESR 1 mm/hour, CRP 0.2 mg/dL), coinciding with complete recovery of right third cranial nerve function. This favorable parallel clinical and biochemical evolution is consistent with previous reports and underscores the benefit of aggressive early immunosuppressive therapy in preventing irreversible ischemic complications [2,8]. Although adjunctive steroid-sparing agents such as tocilizumab may be considered, especially in refractory cases, they were not required in this instance [19]. Close clinical and laboratory follow-up is essential to monitor treatment response and taper regimens judiciously [18,19].
Conclusions
This case illustrates a rare presentation of GCA manifesting as third cranial nerve palsy with ptosis and concurrent CNS vasculitis. Clinicians should maintain a high index of suspicion for GCA in older adults with an abrupt onset of diplopia, even when AION is absent. Prompt recognition, thorough diagnostic workup, and immediate initiation of high-dose corticosteroid therapy are essential to prevent severe complications, most importantly permanent vision loss. Particularly, the association between GCA and findings suggestive of CNS vasculitis is exceptionally rare, further contributing to the uniqueness of this case. Further research is needed to clarify the prevalence and prognostic implications of CNS vasculitis in GCA.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Giant cell arteritis: early diagnosis is key Eye Brain Baig IF Pascoe AR Kini A Lee AG 112112019 https://pmc.ncbi.nlm.nih.gov/articles/PMC 6340646/3069709210.2147/EB.S 170388 PMC 6340646 · doi ↗ · pubmed ↗
- 2Ocular manifestations of giant cell arteritis Am J Ophthalmol Hayreh SS Podhajsky PA Zimmerman B 5095201251998 https://www.ajo.com/article/S 0002-9394(99)80192-5/abstract 955973710.1016/s 0002-9394(99)80192-5 · doi ↗ · pubmed ↗
- 3Ophthalmic manifestations of giant cell arteritis Rheumatology (Oxford) Vodopivec I Rizzo JF 3rd 072572018 https://pubmed.ncbi.nlm.nih.gov/29986083/10.1093/rheumatology/kex 42829986083 · doi ↗ · pubmed ↗
- 4Retrospective, multicenter comparison of the clinical presentation of patients presenting with diplopia from giant cell arteritis vs other causes J Neuroophthalmol Ross AG Jivraj I Rodriguez G 813392019 https://journals.lww.com/jneuro-ophthalmology/fulltext/2019/03000/retrospective,_multicenter_comparison_of_the.3.aspx 2969744110.1097/WNO.0000000000000656 · doi ↗ · pubmed ↗
- 5Diagnosis and classification of central nervous system vasculitis J Autoimmun Hajj-Ali RA Calabrese LH 14915248-492014 https://www.sciencedirect.com/science/article/abs/pii/S 0896841114000092?via%3Dihub 2449182210.1016/j.jaut.2014.01.007 · doi ↗ · pubmed ↗
- 62022 American College of Rheumatology/EULAR classification criteria for giant cell arteritis Arthritis Rheumatol Ponte C Grayson PC Robson JC 18811889742022 https://onlinelibrary.wiley.com/doi/full/10.1002/art.423253635012310.1002/art.42325 · doi ↗ · pubmed ↗
- 7Trial of tocilizumab in giant-cell arteritis N Engl J Med Stone JH Tuckwell K Dimonaco S 3173283772017 https://www.nejm.org/doi/full/10.1056/NEJ Moa 16138492874599910.1056/NEJ Moa 1613849 · doi ↗ · pubmed ↗
- 8Population-based rate and patterns of diplopia in giant cell arteritis Neuroophthalmology Castillejo Becerra CM Crowson CS Koster MJ Warrington KJ Bhatti MT Chen JJ 7579462022 https://pubmed.ncbi.nlm.nih.gov/35273408/3527340810.1080/01658107.2021.1965627 PMC 8903753 · doi ↗ · pubmed ↗