Germline variants in ATM, BRCA2, other cancer predisposition and novel candidate genes are implicated in glioma risk in adult glioma patients with a familial or personal history of tumors
Frank Brand, Lily S. Rose, Amir H. Akbarzadeh, Christine A. M. Weber, Isabel Eckert, Gunnar Schmidt, Bernd Auber, Alisa Förster, Ulrike Beyer, Robert Geffers, Stephan Bartels, Michael Lalk, Manolis Polemikos, Michael Friese, Michael Sabel, Philipp Schwenkenbecher, Paul Kremer

TL;DR
This study finds that germline variants in cancer-related genes like ATM and BRCA2 are linked to glioma risk in patients with a family or personal tumor history.
Contribution
The study identifies novel and known cancer predisposition genes associated with glioma risk in patients with tumor histories.
Findings
Germline variants in DNA damage response genes like ATM and BRCA2 were found in 23% of glioma patients with tumor histories.
ATM variants were linked to younger diagnosis age and IDH-mutant astrocytoma, while BRCA2 variants were enriched in glioma patients.
Some patients with germline variants may benefit from targeted therapies like PARP or immune checkpoint inhibitors.
Abstract
Familial occurrence of gliomas has been reported in around 5% of patients. Studies on the genetic landscape of glioma predisposition are scarce. Here, leukocyte DNA of 213 adult glioma patients with a familial and/or personal tumor history from 206 families was subjected to whole-exome sequencing. Germline variants (GVs) were analyzed using two approaches: (1) GVs in 164 established cancer predisposition genes (CPGs) or suspected glioma risk genes were extracted and classified; (2) the enrichment of genes with loss-of-function or deleterious missense GVs that were ultrarare or ClinVar likely pathogenic/pathogenic in the glioma versus a control cohort (n = 391) was determined. In 23% (48/213) of glioma patients with a familial/personal tumor history, GVs predicted to be deleterious in CPGs were detected. Of the mutated CPGs, 37% were involved in DNA damage response, including ATM, BRCA2,…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
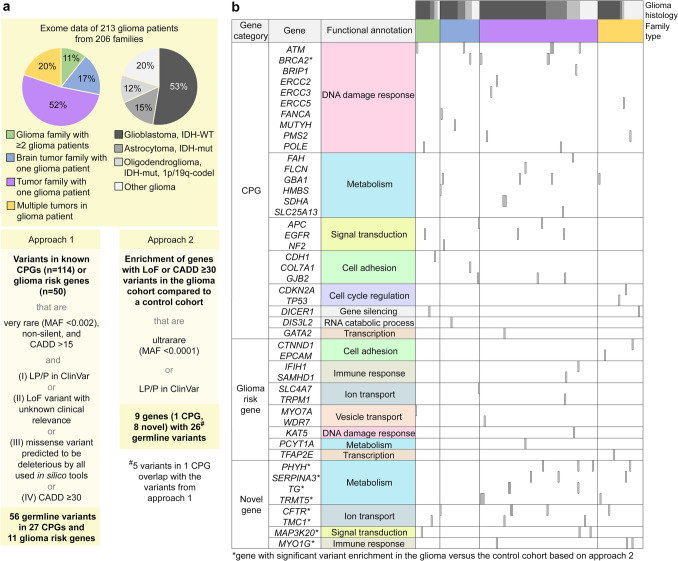 Figure 1
Figure 1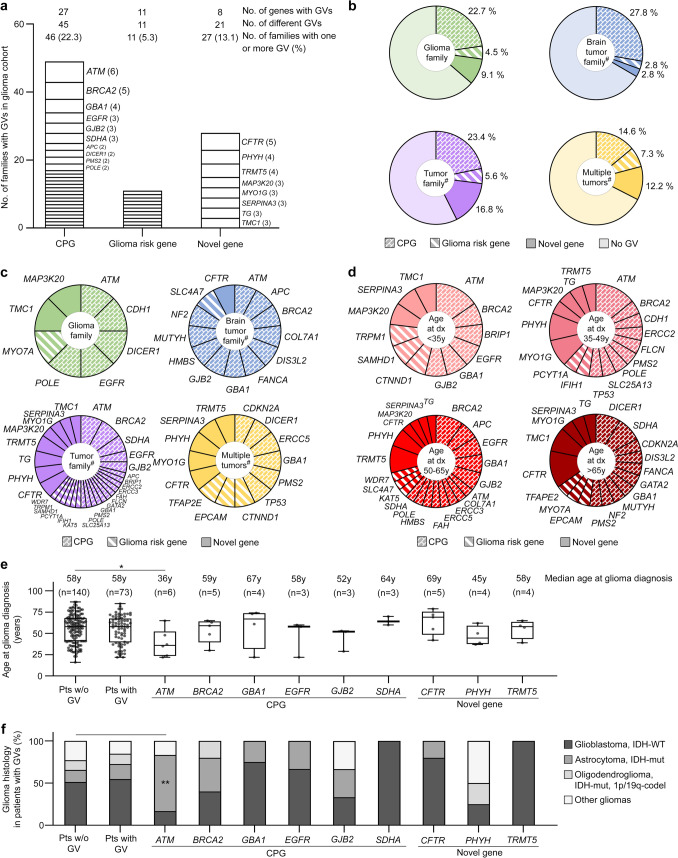 Figure 2
Figure 2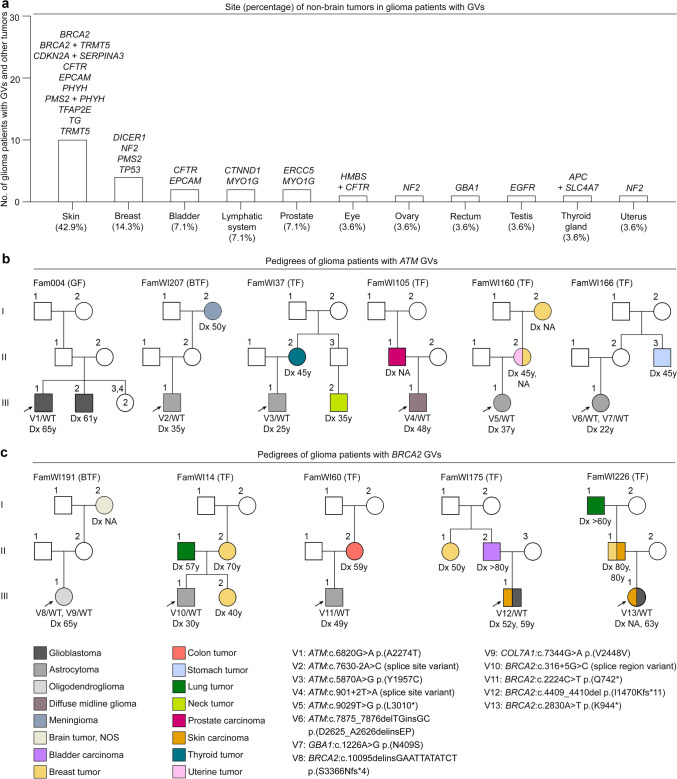 Figure 3
Figure 3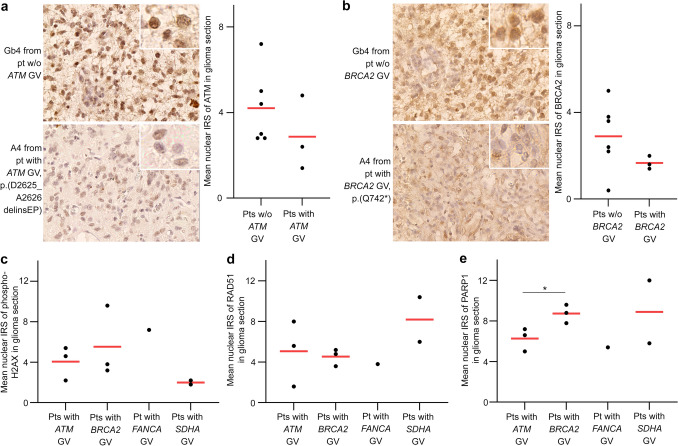 Figure 4
Figure 4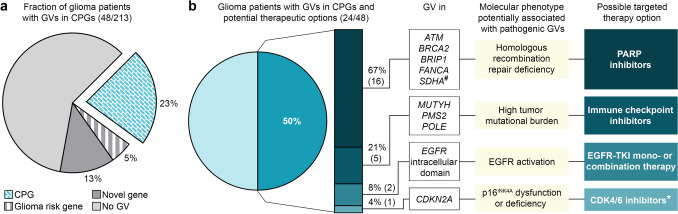 Figure 5
Figure 5- —Förderstiftung MHH plus
- —https://doi.org/10.13039/100008672Wilhelm Sander-Stiftung
- —Medizinische Hochschule Hannover (MHH) (3118)
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsGlioma Diagnosis and Treatment · Cancer Genomics and Diagnostics · Genomics and Rare Diseases
Introduction
Adult-type diffuse gliomas represent a group of primary brain tumors that comprises IDH-mutant astrocytoma, IDH-mutant and 1p/19q-codeleted oligodendroglioma, and IDH-wildtype glioblastoma according to the 2021 World Health Organization (WHO) Classification of Tumors of the Central Nervous System (CNS) [45]. Although most primary brain tumors, including gliomas, occur sporadically, familial aggregation of primary brain tumors has been reported in approximately 5% of patients [47, 81]. A number of monogenic autosomal-dominant hereditary tumor syndromes, such as Lynch, Li-Fraumeni, and melanoma-astrocytoma syndrome, caused by heterozygous pathogenic germline variants in cancer predisposition genes (CPGs), such as PMS2, TP53, and CDKN2A, are associated with an increased risk of gliomas [34, 37]. Some autosomal-recessive tumor predisposition syndromes, such as ataxia telangiectasia, caused by biallelic variants in the ATM gene, have also been linked to glioma predisposition [37].
Recently, first attempts have been made to determine the prevalence and type of pathogenic germline variants in adult glioma using next-generation sequencing approaches, including targeted exome and whole-genome sequencing [15, 32, 51]. These studies investigated unselected glioma patients presenting at defined centers in the United States over a certain time period [32, 51], or glioma families with at least two members diagnosed with glioma each, recruited at 14 centers in the United States, Israel, Sweden, and Denmark [15], and identified known and novel glioma risk genes and variants therein. To further elucidate the genetic landscape of glioma predisposition, we performed whole-exome sequencing (WES) on leukocyte DNA of 213 adult glioma patients diagnosed and treated in Germany with a familial and/or personal medical history of tumors. Data were analyzed using a candidate gene approach taking established CPGs and suspected glioma risk genes into account (approach 1), and a comparison of genetic findings was done in patients and controls (approach 2). We present detailed genotype–phenotype data for each carrier of a pathogenic germline variant (GV) in CPGs and glioma risk genes and the respective family, explore the resulting pathomechanisms of glioma tumorigenesis, and discuss potential therapeutic options for GV carrying patients.
Materials and methods
Human samples
The study was approved by the ethics boards of the participating centers in Hannover, Germany. Each family provided informed consent for participation in the study. The glioma cohort consisted of 206 families with at least one glioma patient each and presumed tumor predisposition due to (i) at least one additional glioma patient in the family (glioma family, n = 22, 11%), (ii) at least one additional brain tumor patient not otherwise specified in the family (brain tumor family, n = 36, 17%), (iii) at least one additional non-brain tumor patient in the family (tumor family, n = 107, 52%), or (iv) a glioma patient with a personal medical history of at least one syn- or metachronous non-brain tumor with an unremarkable family history (multiple tumors, n = 41, 20%). In the 206 families, blood samples were available from 213 patients diagnosed with a primary glioma at a median age of 58 years, who underwent brain tumor surgery during the time period December 2012–February 2024, mostly in Hannover, Germany. Tumor classification was originally done according to the WHO Classification of Tumours of the CNS of 2007, 2016, or 2021. In this study, the 213 primary gliomas were divided into four tumor types based on the WHO Classification of Tumours of the CNS of 2021: glioblastoma, IDH-wildtype (n = 112, 53%), astrocytoma, IDH-mutant (n = 33, 15%), oligodendroglioma, IDH-mutant and 1p/19q-codeleted (n = 25, 12%), or, if they did not fit unequivocally into the former three groups, as other gliomas (n = 43, 20%).
The control cohort used in approach 2 consisted of 391 in-house control individuals with no personal tumor history. These samples were chosen because they were from non-tumor individuals recruited at the same center and during the same time period as the glioma cohort, and sequencing and processing were identical. Control samples underwent WES concurrently with glioma samples and were analyzed in an identical manner.
DNA extraction and WES
DNA was isolated from peripheral blood using the QIAamp DNA Blood Maxi Kit (Qiagen, Hilden, Germany) and from formalin-fixed paraffin-embedded (FFPE) glioma tissue using the QIAamp DNA FFPE Advanced Kit (Qiagen). WES was performed on leukocyte DNA of 213 glioma patients and 391 control individuals, and on tumor DNA of five glioma patients carrying BRCA2 or MUTYH GVs using the SureSelectXT Human All Exon (Agilent, Santa Clara, CA, USA) or IDT xGen Exome Research Panel v2 (Integrated DNA Technologies, Coralville, IA, USA) target enrichment kit on a HiSeq or NovaSeq sequencer (all Illumina, San Diego, CA, USA). All samples were sequenced to a mean target coverage of ≥ 50x (leukocyte DNA) or > 200x (tumor DNA).
WES data analysis
Sequencing data were aligned to the human reference genome build hg38/GRCh38, and variations were called using CLC Genomic Workbench (version 24.0.2; Qiagen). Variations were annotated and prioritized using Clinical Insight Interpret Translational (Qiagen). Quality filters were applied (coverage ≥ 20, call quality ≥ 25, allele fraction ≥ 30) and non-silent variants (splice site region up to 5 bases into intron, frameshift, in-frame indel (only in approach 1), stop gain/loss, and non-synonymous missense variants) were retained. Variant minor allele frequencies (MAF) for the non-Finnish European population were retrieved from the Genome Aggregation Database browser v4.1.0 (https://gnomad.broadinstitute.org). For prediction of variant deleteriousness, the tools CADD (https://cadd.gs.washington.edu), SIFT (https://sift.bii.a-star.edu.sg), PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2), FATHMM Cancer (http://fathmm.biocompute.org.uk/cancer.html), and the metapredictor REVEL were used. An effect of variants on splicing was predicted by SpliceAI (https://spliceailookup.broadinstitute.org/) and MaxEntScan (https://github.com/matthdsm/MaxEntScan). Clinical classification of variants was retrieved from the ClinVar database (https://www.ncbi.nlm.nih.gov/clinvar).
Two approaches were used to prioritize the identified GVs. In approach 1, we only considered GVs in 114 established CPGs according to Rahman 2014 [63], and 50 potential glioma risk genes according to Choi et al. [15] and others (genes and references are listed in Supplementary Table 1 online resource) that were very rare (MAF < 0.002), had a CADD score > 15, and were (i) likely pathogenic or pathogenic (LP/P) according to the ClinVar database, (ii) predicted loss-of-function (LoF) variants with conflicting pathogenicity in ClinVar or not listed, (iii) missense variants predicted to be deleterious by all in silico tools used (CADD, SIFT, PolyPhen-2, REVEL, and FATHMM Cancer), or (iv) had a high CADD score (≥ 30), but did not fulfill criteria (i), (ii) or (iii) (Fig. 1a). For these GVs, the term pathogenic GVs was used. In approach 2, we identified genes with an enrichment of LoF variants (including start-loss, frameshift, splice region (± 5) with an effect on splicing according to MaxEntScan, and stop-gain variants, excluding in-frame indels) and/or of non-silent variants with a CADD score ≥ 30 (equivalent to the top 0.1% most deleterious variants) that were ultrarare (MAF < 0.0001) or classified as LP/P (ClinVar, any level of evidence) in the germline of the glioma (n = 206 families) compared to the control (n = 391 families) cohort. Only unrelated genetic data, i.e., from one glioma patient per family, were used, and only genes with no variants specified above in the control cohort and ≥ 3 variants specified above in the glioma cohort were considered in approach 2. Genes with GVs identified by approach 1 and 2 were functionally annotated using the web tool DAVID (https://davidbioinformatics.nih.gov).Fig. 1. Whole-exome sequencing to identify genetic determinants of glioma predisposition: study design with an overview of diagnostic yield, and landscape of pathogenic germline variants and affected genes. a Scheme describing the family type and glioma histology of the study cohort and the two approaches used for analysis of WES data with an overview of the diagnostic yield. b Landscape of pathogenic germline variants in CPGs, suspected glioma risk genes, and novel candidate genes sorted by gene category, functional annotation, and family type. Glioma histology is also indicated. CADD combined annotation dependent depletion score; ClinVar ClinVar database; codel codeleted; CPG cancer predisposition gene; IDH isocitrate dehydrogenase 1/2; LoF loss-of-function; LP likely pathogenic; MAF minor allele frequency; mut mutant; P pathogenic; WT wildtype
Expression analysis in glioma sections by immunohistochemistry
Immunohistochemical staining of ATRX, IDH1 R132H, and p53 was done as previously described [30]. The expression of ATM, BRCA2, phospho-H2AX, RAD51, and PARP1 was analyzed on FFPE glioma sections from patients with GVs in ATM, BRCA2, FANCA, SDHA after heat-induced epitope retrieval in 10 mM citrate buffer pH 6.0. The primary antibodies used were: rabbit anti-ATM (1:40 dilution; ab32420, Abcam, Cambridge, United Kingdom), mouse anti-BRCA2 (1:40 dilution; MAB2476, R&D Systems, Inc., Minneapolis, MN, USA), rabbit anti-phospho-H2AX (Ser139) (1:100 dilution; #9718, Cell Signaling Technologies, Danvers, MA, USA), rabbit anti-RAD51 (1:400 dilution; GTX100469, GeneTex, Irvine, CA, USA), and rabbit anti-poly(ADP-ribose) polymerase 1 (PARP1) (1:50 dilution; ab6079, Abcam). As secondary antibodies, horseradish peroxidase-labeled donkey anti-mouse or anti-rabbit IgG (1:300 dilution; A16017 / A16035, Thermo Fisher Scientific, Waltham, MA, USA) were used. Sections were counterstained with Mayer’s hemalum solution (Carl Roth GmbH + Co. KG, Karlsruhe, Germany), scanned using the Metafer Scanning Platform (MetaSystems Hard & Software, Altlussheim, Germany), and digital images were processed using ImageScope v11.2.0.780 software (Leica Microsystems, Wetzlar, Germany). The nuclear staining was assessed in five non-overlapping fields per section, and the immunoreactivity score (IRS) was determined. The IRS is the product of a proportion score (0: 0%, 1: < 10%, 2: 10–50%, 3: 50–80%, 4: > 80% positive cells) and an intensity score (0: negative, 1: weak, 2: moderate, 3: strong reaction) with a range from 0 to 12. The mean IRS was calculated from five fields per section (0–1: negative, 2–3: weak, 4–8: moderate, 9–12: strong expression).
The expression of EGFR and phospho-EGFR (Tyr1068) was analyzed on FFPE tumor sections from glioblastoma patients with GVs in EGFR after heat-induced epitope retrieval in Cell Conditioning 1 (Roche, Basel, Switzerland) or in 10 mM citrate buffer pH 6.0. The primary antibodies used were: rabbit anti-EGFR (undiluted, 0.4 µg/ml; 790–4347, Roche) and rabbit anti-phospho-EGFR (Tyr1068) (1:100 dilution; #3777, Cell Signaling Technologies). As secondary antibodies, ultraView Universal HRP Multimer (undiluted; included in the ultraView Universal DAB Detection Kit, 760–500, Roche) or horseradish peroxidase-labeled donkey anti-rabbit IgG (1:300 dilution; A16035, Thermo Fisher Scientific) were used. Sections were counterstained with Mayer’s hemalum solution (Carl Roth GmbH + Co. KG). For histological evaluation, consecutive sections were stained with hematoxylin-eosin according to standard protocols. Images were acquired using a BX46 microscope and a XC50 camera (all Olympus, Shinjuku, Japan).
Molecular glioma characterization
Variant hotspot sequencing of the IDH1 and IDH2 genes was done, and MGMT promoter methylation was determined according to previously reported protocols [16]. Fluorescence in situ hybridization of the EGFR locus, variant hotspot sequencing of the H3F3A, HIST1H3B, and TERT genes, and digital PCR to assess CDKN2A/B deletion were reported in detail elsewhere [30]. To determine the tumor mutational burden (TMB), glioma tissue was microdissected from 5 µm FFPE tumor sections. DNA was extracted using the Maxwell RSC DNA FFPE Kit (Promega, Madison, WI, USA) and a Maxwell RSC Instrument (Promega). For TMB detection, 15 ng tumor DNA was used in an Oncomine Tumor Mutation Load Assay (n = 409 genes, 1.7 Mb exonic coverage, Thermo Fisher Scientific) on an Ion S5 Prime System (Life Technologies, Carlsbad, CA, USA). For TMB calculation, quality and population filters were applied (coverage ≥ 60, strand bias ≥ 0.9, MAF < 0.05). Second hit analysis was based on WES data obtained from tumor DNA of glioma patients carrying BRCA2 or MUTYH GVs, taking variants with a MAF < 0.01 into account.
Statistical analysis
Statistical analysis was conducted using MATLAB (The MathWorks, Natick, MA, USA) or Prism software (GraphPad Software, Boston, MA, USA) and Mann–Whitney test or Fisher’s exact test (both two-tailed). P values < 0.05 were considered significant. Multiple testing in approach 2 was adjusted for with a false discovery rate of 10% according to Benjamini, Krieger, and Yekutieli.
Results
Rare deleterious variants in CPGs or suspected glioma risk genes in the germline of glioma patients with presumed tumor predisposition
To explore the role of GVs in glioma risk, we analyzed germline WES data of 213 glioma patients from 206 families with evidence for a tumor predisposition using two approaches (Fig. 1a). In approach 1, we prioritized very rare (MAF < 0.002) GVs with a CADD score > 15 in 114 established CPGs [24, 63, 80] and 50 potential glioma risk genes identified by us or others [3, 6, 15, 18] (genes and references listed in Supplementary Table 1 online resource). Of the detected 348 GVs, 20 were ClinVar LP/P, 10 were predicted LoF variants with conflicting pathogenicity in ClinVar or not listed, 18 were missense variants predicted to be deleterious by all in silico tools used (CADD, SIFT, PolyPhen-2, REVEL, FATHMM Cancer), and eight had a high CADD score (≥ 30), but did not fulfill the other criteria (Table 1). Taken together, 56 GVs in 27 CPGs and 11 suspected glioma risk genes were considered pathogenic GVs (Fig. 1a), and 56 glioma patients from 54 families were affected by at least one pathogenic GV in a CPG and/or glioma risk gene (Supplementary Tables 2a, 2b online resource). The CPGs affected by pathogenic GVs were most frequently associated with DNA damage response (e.g., ATM, BRCA2, PMS2, POLE), metabolism (e.g., GBA1, SDHA), signal transduction (e.g., APC, EGFR), and cell adhesion (e.g., CDH1, GJB2) (Fig. 1b). Potential glioma risk genes with a pathogenic GV most frequently played roles in cell adhesion (CTNND1, EPCAM), immune response (IFIH1, SAMHD1), and ion transport (SLC4A7, TRPM1) (Fig. 1b).Table 1. Heterozygous germline variants identified in cancer predisposition genes or suspected glioma risk genes in 213 adult glioma patients with presumed tumor predisposition from 206 families using approach 1Patient IDGeneInheritance^a^AD inheritance previously observed^b^Genomic position (GRCh38/hg38)Nucleotide changeProtein/molecular consequenceClin Var^c^MAF^d^(%)Prediction according to CADD^e^/SIFT^f^/PolyPhen-2^ g^/REVEL^h^/FATHMM Cancer^i^/SpliceAI^j^Likely pathogenic or pathogenic variants according to ClinVarWI207-III.1ATMAD/AR + 11:108,331,877c.7630-2A>CSplice acceptorP0.00144134.0/—/—/—/—/0.91WI166-III.1ATMAD/AR + 11:108,332,848c.7875_7876delinsGCp.(D2625_A2626delinsEP)LP/P022.4/—/—/—/—/—WI14-III.1BRCA2AD/AR + 13:32,319,330c.316+5G>CSplice regionP022.3/—/—/—/—/0.92WI60-III.1BRCA2AD/AR + 13:32,336,579c.2224C>Tp.(Q742*)P033.0/—/—/—/—/—WI226-III.1BRCA2AD/AR + 13:32,337,185c.2830A>Tp.(K944*)P0.00033932.0/—/—/—/—/—WI175-III.1BRCA2AD/AR + 13:32,338,763c.4409_4410delp.(I1470Kfs11)P021.2/—/—/—/—/—WI86-III.1BRIP1AD/AR + 17:61,859,791c.205+5G>TSplice regionLP024.2/—/—/—/—/0.85WI04-III.1CDKN2AAD + 9:21,971,200c.159G>Ap.(M53I)LP/P027.1/D/B/0.724/−2.86/0.02WI191-III.1COL7A1AD/AR + 3:48,570,639c.7344G>Ap.(V2448V) splice donorP0.00663822.8/—/—/—/—/0.63WI165-III.1EPCAMAD/AR + 2:47,374,035c.412C>Tp.(R138)P0.000169542.0/—/—/—/—/—WI50-III.1FAHAR − 15:80,180,230c.1062+5G>ASplice regionP0.0464223.5/—/—/—/—/ 0.78WI209-III.1FANCAAR(+)^k,l^16:89,746,848c.3391A>Gp.(T1131A)LP/P0.0161425.5/D/PoD/0.842/3.74/0.09WI166-III.1WI205-III.1WI214-III.1GBA1AD/AR − 1:155,235,843c.1226A>Gp.(N409S)LP/P0.172824.1/D/B/0.673/2.36/0.21WI09-III.1GBA1AD/AR − 1:155,235,195c.1505G>Ap.(R502H)LP/P0.000339134.0/D/PrD/0.842/2.32/0.38WI75-III.1GJB2AD/AR + 13:20,189,117c.465T>Ap.(Y155*)P0.0000847429.5/—/—/—/—/—WI48-III.1WI49-III.1GJB2AD/AR + 13:20,189,473c.109G>Ap.(V37I)P0.0881621.7/T/PrD/0.656/3.08/0.02WI70-III.1MUTYHAR(+)^m,n^1:45,332,445c.650G>Ap.(R217H)LP/P0.00669525.8/D/PrD/0.927/0.27/0.08Fam003-III.1MYO7AAD/AR(+)^o^11:77,184,688c.3476G>Tp.(G1159V)LP/P0.0535529.1/D/PrD/0.938/0.53/0.00WI78-III.1SAMHD1AD/AR − 20:36,951,576c.68C>Gp.(S23*)LP/P0.00254233.0/—/—/—/—/—LI06-III.1WI87-III.1SDHAAD/AR + 5:223,509c.91C>Tp.(R31*)LP/P0.0534134.0/—/—/—/—/—Predicted loss-of-function variants with conflicting pathogenicity in ClinVar or not listedWI105-III.1ATMAD/AR + 11:108,245,028c.901+2T>ASplice donor–033.0/—/—/—/—/0.99WI160-III.1ATMAD/AR + 11:108,365,366c.9029T>Gp.(L3010*)–042.0/—/—/—/—/—WI191-III.1BRCA2AD/AR + 13:32,398,608c.10095delinsGAATTATATCT p.(S3366Nfs4)C024.2/—/—/—/—/—WI72-III.1ERCC5AR − 13:102,854,319c.412C>Tp.(R138)–0.00033936.0/—/—/—/—/—WI53-III.1KAT5AD − 11:65,718,667c.1342C>Tp.(R448*)–0.000169537.0/—/—/—/—/—WI222-III.1NF2AD + 22:29,694,776c.1762C>Tp.(R588*)VUS0.00067840.0/—/—/—/—/—WI22-III.2PCYT1AAR − 3:196,238,789c.1003C>Tp.(R335*)VUS0.00102840.0/—/—/—/—/—WI88-III.1POLEAD/AR + 12:132,687,315c.1A>T (start codon variant)p.(M1L)C0.0149521.9/D/B/0.257/3.37/0.01WI183-III.1SDHAAD/AR + 5:254,390c.1795-3C>GSplice regionC0.000170624.1/—/—/—/—/0.85WI48-III.1TRPM1AR(+)^p^15:31,002,524c.4173_4176delAGACp.(D1392Lfs11)VUS0.0166925.0/—/—/—/—/—Missense variants with pathogenic predictions in all used in silico tools^q^WI99-III.1APCAD + 5:112,841,473c.5879C>Tp.(P1960L)C0.00534125.4/D/PrD/0.878/−5.39/0.00WI89-III.1APCAD + 5:112,842,085c.6491G>Tp.(G2164V)–026.2/D/PrD/0.558/−4.46/0.00WI37-III.1ATMAD/AR + 11:108,310,267c.5870A>Gp.(Y1957C)VUS0.000169526.9/D/PrD/0.602/−2.22/0.01Fam004-III.1ATMAD/AR + 11:108,326,070c.6820G>Ap.(A2274T)C0.0182227.5/D/PoD/0.524/−0.94/0.02Fam002^r^-II.2-III.1,-III.2CDH1AD + 16:68,833,300c.2450C>Tp.(A817V)C0.00211927.2/D/PrD/0.574/−1.22/0.03WI239-III.1DICER1AD + 14:95,091,226c.5504A>Gp.(Y1835C)C0.00457628.8/D/PoD/0.736/−1.05/0.15WI61-III.1DICER1AD + 14:95,107,993c.2537T>Gp.(I846S)–024.5/D/PoD/0.739/−1.63/0.07WI201-III.1EGFRAD/AR + 7:55,154,015c.752G>Cp.(C251S)–027.7/D/PrD/0.876/−3.48/0.02WI177-II.1EGFRAD/AR + 7:55,174,726c.2189T>Gp.(L730R)C0.00220431.0/D/PrD/0.573/−1.9/0.02WI33-III.1EGFRAD/AR + 7:55,200,352c.2885G>Ap.(R962H)C0.0222929.5/D/PoD/0.543/−1.97/0.07WI153-III.1ERCC2AR(+)^s^19:45,352,511c.2041G>Ap.(D681N)C0.00169524.9/D/PrD/0.898/−1.48/0.05WI22-III.2FLCNAD + 17:17,216,464c.1216A>Gp.(S406G)C0.00440725.3/D/PoD/0.672/−2.08/0.04LI06-III.1GATA2AD + 3:128,487,002c.30G>Tp.(W10C)C0.0188132.0/D/PoD/0.947/−1.25/0.00WI169-II.2HMBSAD/AR + 11:119,088,642c.95G>Ap.(R32H)VUS0.000169629.2/D/PrD/0.885/NA/0.06WI11-III.1PMS2AD/AR + 7:5,992,044c.917T>Cp.(V306A)VUS0.00191627.6/D/PrD/0.824/−1.66/0.00WI103-III.1PMS2AD/AR + 7:5,995,563c.874A>Tp.(I292F)VUS0.000254325.7/D/PrD/0.866/−1.88/0.06WI51-III.1TFAP2E*–(+)^t^1:35,590,037c.893C>Tp.(S298L)VUS0.00898428.3/D/PoD/0.922/−1.28/0.01WI236-III.1TP53AD + 17:7,675,220c.392A>Gp.(N131S)VUS0.0000847527.0/D/PrD/0.859/−9.14/0.00Variants with a CADD score ≥ 30, but not fulfilling the other criteriaWI122-III.1CTNND1AD(+)^u^11:57,814,325c.2653C>Tp.(R885W)VUS0.00186732.0/D/PoD/0.414/2.28/0.12WI163-III.1DIS3L2AR(+)^v^2:232,136,590c.821G>Ap.(R274Q)VUS031.0/D/PrD/0.497/2.45/0.03WI126-II.2ERCC3AR(+)^w^2:127,286,825c.1220T>Gp.(I407S)–030.0/D/PrD/0.710/1.87/0.00WI106-III.1IFIH1AD/AR − 2:162,277,667c.1792C>Tp.(R598C)VUS0.00314832.0/D/PrD/0.556/2.27/0.00Fam016-III.1POLEAD/AR + 12:132,657,402c.3406C>Tp.(R1136W)VUS0.000423730.0/D/PrD/0.598/0.57/0.00WI89-III.1SLC4A7– − 3:27,394,756c.2879A>Gp.(Y960C)–0.000423832.0/D/PrD/0.939/3.01/0.00WI145-III.1SLC25A13AR(+)^x^7:96,189,581c.848G>Ap.(G283E)VUS0.00908633.0/D/B/0.709/3.42/0.72WI08-III.1WDR7– − 18:56,816,031c.3191C>Tp.(P1064L)VUS0.000512433.0/D/PoD/0.242/0.01/0.00Listed are very rare (MAF < 0.002), non-silent (i.e., splice site, frameshift, in-frame indels, stop gain/loss and non-synonymous missense) variants with a CADD score > 15, classified as LP/P in ClinVar, predicted loss-of-function variants with conflicting pathogenicity in ClinVar or not listed, missense variants with pathogenic predictions in all used in silico tools, or with a CADD score ≥ 30, but not fulfilling the other criteriaAD autosomal dominant; AR autosomal recessive; C conflicting classifications of pathogenicity in the ClinVar database; delins deletion insertion; D damaging; fs frameshift; LP likely pathogenic; MAF minor allele frequency; P pathogenic; PoD possibly damaging; PrD probably damaging; T tolerated; VUS variant of uncertain significance; *, stop gain; –, no data^a^Inheritance according to the OMIM database (https://www.omim.org)^b^Autosomal dominant inheritance observed in cancer patients according to Rahman 2014 [63] (indicated by +) or the reports cited in the footnotes^c^Classification according to the ClinVar database (www.ncbi.nlm.nih.gov/clinvar)^d^Minor allele frequency according to the Genome Aggregation Database (gnomAD) browser version 4.1.0, non-Finnish European population (https://gnomad.broadinstitute.org)^e^According to CADD (https://cadd.gs.washington.edu)^f^According to SIFT (https://sift.bii.a-star.edu.sg)^g^According to PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2)^h^According to REVEL (https://sites.google.com/site/revelgenomics/downloads/revel-genome-segment-files)^i^According to Functional Analysis through Hidden Markov Models (FATHMM) version v2.3 for cancer-promoting/driver mutations and other germline polymorphisms (http://fathmm.biocompute.org.uk/cancer.html)^j^According to SpliceAI (https://spliceailookup.broadinstitute.org)^k^Autosomal dominant inheritance observed in cancer patients[71]^l^Autosomal dominant inheritance observed in cancer patients[1]^m^Autosomal dominant inheritance observed in cancer patients[4]^n^Autosomal dominant inheritance observed in cancer patients[36]^o^Autosomal dominant inheritance observed in cancer patients[23]^p^Autosomal dominant inheritance observed in cancer patients[79]^q^Variants with CADD score ≥ 20; FATHMM Cancer ≤ −0.75; REVEL score > 0.5; predicted to be damaging in SIFT and PolyPhen-2^r^This family was previously published[24]^s^Autosomal dominant inheritance observed in cancer patients[82]^t^Autosomal dominant inheritance observed in cancer patients[12]^u^Autosomal dominant inheritance observed in cancer patients[29]^v^Autosomal dominant inheritance observed in cancer patients[25]^w^Autosomal dominant inheritance observed in cancer patients[74]^x^Autosomal dominant inheritance observed in cancer patients[14]
Genes enriched with LoF or deleterious missense GVs in glioma patients with presumed tumor predisposition compared to a control cohort
In approach 2, we searched for genes significantly more frequently affected by LoF variants (excluding in-frame indels) or missense variants with a CADD score ≥ 30 that were ultrarare (MAF < 0.0001) or ClinVar LP/P in the glioma cohort, i.e., glioma index patients from 206 families, versus a control cohort, i.e., control individuals from 391 families. After adjustment for multiple testing with a false discovery rate of 10%, a significant GV enrichment in the glioma versus the control cohort was found for nine genes, i.e., one CPG (BRCA2) and eight novel genes not previously associated with glioma risk, with 26 different GVs considered pathogenic GVs (Fig. 1a, Supplementary Table 3 online resource). Pathogenic GVs in BRCA2 were detected in five families of the glioma cohort but not in controls (P = 0.005), implicating BRCA2 using both approaches utilized here. The eight novel genes with pathogenic GVs in three to five families from the glioma cohort but not in controls (P < 0.05) play roles in metabolism (PHYH, SERPINA3, TG, TRMT5), ion transport (CFTR, TMC1), immune response (MYO1G), and signal transduction (MAP3K20) (Fig. 1b, Supplementary Table 3 online resource).
All genetic determinants of glioma predisposition identified in glioma patients with presumed tumor predisposition
When combining the results from approach 1 (Table 1) and from approach 2 (Supplementary Table 3 online resource), CPGs were affected by at least one pathogenic GV in 46/206 (22.3%) families, suspected glioma risk genes in 11/206 (5.3%) families, and novel genes with significant GV enrichment in 27/206 (13.1%) families of the glioma cohort (Fig. 2a). The CPGs most frequently affected by GVs were ATM in 6/206 (2.9%) families, BRCA2 in 5/206 (2.4%) families (identified in approaches 1 and 2), GBA1 in 4/206 (1.9%) families as well as EGFR, GJB2, and SDHA in 3/206 (1.5%) families each of the glioma cohort (Fig. 2a). No potential glioma risk gene was recurrently affected by GVs. The novel genes most frequently affected by GVs were CFTR in 5/206 (2.4%) families followed by PHYH and TRMT5 in 4/206 (1.9%) families each of the glioma cohort (Fig. 2a, Supplementary Table 3 online resource). While the fraction of pathogenic GVs in CPGs was similar in glioma families with at least two glioma patients each as well as in brain tumor families and tumor families with at least one glioma patient each (22.7%, 27.8%, and 23.4%), it was lower in multiple tumor glioma patients who had a personal medical history of at least one syn- or metachronous non-brain tumor but an unremarkable family history (14.6%) (Fig. 2b). Pathogenic GVs in ATM were found in each family type with the highest fraction in glioma families, but not in multiple tumor glioma patients (Fig. 2c). Pathogenic GVs in BRCA2 were detected in tumor families and brain tumor families. Pathogenic GVs in suspected glioma risk genes and novel genes were observed in each family type (Fig. 2c).Fig. 2. Whole-exome sequencing results in 213 glioma patients with a familial and/or personal history of tumors from 206 families obtained by approach 1 and 2, and characteristics of 73 glioma patients with pathogenic GVs. a Number of glioma families with pathogenic GVs in CPGs, suspected glioma risk genes, and novel candidate genes. Genes recurrently affected are specified, and the number of families with a GV in a certain gene is given in brackets. b Diagnostic yield of pathogenic GVs in CPGs, potential glioma risk genes, and novel candidate genes in the four types of families with one glioma patient each and at least another glioma patient (glioma family), brain tumor patient (brain tumor family), tumor patient (tumor family) or no family history of tumors but syn-/metachronous non-brain tumors in the personal history of the glioma patient (multiple tumors). c Genes (CPGs, suspected glioma risk genes, and novel candidate genes) affected by pathogenic GVs in the four types of families with at least one glioma patient each. In the pie chart referring to tumor families, the size of each segment reflects the number of families with a GV in the specified gene. d Genes (CPGs, suspected glioma risk genes, and novel candidate genes) affected by pathogenic GVs in four age groups. In the pie charts, the size of each segment reflects the number of families with a GV in the specified gene. e Age at glioma diagnosis of patients without GVs, with GVs, and with GVs in specific genes. Shown are box plots and data points. The median age at glioma diagnosis of ATM GV carriers was significantly lower than that of patients without GVs. f Glioma histology of patients without GVs, with GVs, and with GVs in specific genes. ATM GV carriers were significantly more frequently diagnosed with IDH-mutant astrocytoma than patients without GVs. ^#^, each family contains one glioma patient. C**odel codeleted; CPG cancer predisposition gene; dx diagnosis of primary glioma; GV germline variant; IDH isocitrate dehydrogenase 1/2; multiple tumors glioma patient with syn-/metachronous non-brain tumor(s) in personal medical history and unremarkable family history; mut mutant; No. number; Pts patients; w/o without; WT wildtype; y years. *, P < 0.05 (two-tailed Mann–Whitney test); **, P < 0.01 (two-tailed Fisher’s exact test)
Age at glioma diagnosis and glioma histology of patients with pathogenic GVs
To compare the age at diagnosis of the primary glioma, the GV carriers were divided into four age groups (Fig. 2d). Five of six ATM GV carriers were diagnosed with a glioma before 50 years of age. BRCA2 GV carriers were most frequently affected by glioma between the age of 50 and 65 years*.* The DICER1 and most of the SDHA and CFTR GV carriers developed glioma after the age of 65 years (Fig. 2d). The median age at glioma diagnosis in GV carriers and in glioma patients without GVs was 58 years (P = 0.726; Fig. 2e). ATM GV carriers had a significantly younger median age at glioma diagnosis compared to non-GV carriers (36 versus 58 years, P = 0.022) (Fig. 2e).
Next, we compared the glioma histology in carriers of GVs in different genes and non-GV carriers. Astrocytoma, IDH-mutant was diagnosed significantly more frequently in glioma patients with ATM GVs than in those without GVs (4/6 versus 20/140, P = 0.007; Fig. 2f). Patients with GVs in SDHA (n = 3) or TRMT5 (n = 4) were exclusively affected by glioblastoma, IDH-wildtype (Fig. 2f).
Other tumors and familial tumor spectrum in glioma patients with pathogenic GVs
Syn- or metachronous tumors in other organs were diagnosed in 21/73 (28.8%) glioma patients with a pathogenic GV, recurrently affecting the skin, breast, bladder, lymphatic system, and prostate (Fig. 3, Supplementary Figs. 1–7 online resource, Supplementary Table 2a online resource). For example, two glioma patients with a BRCA2 LoF GV also had skin tumors, and both glioma patients with a PMS2 missense GV had a syn- or metachronous tumor of the skin or the breast (Fig. 3a, Fig. 3c, Supplementary Fig. 3 online resource). The carrier of an EPCAM LoF GV had two metachronous tumors affecting the skin and the bladder (Fig. 3a, Supplementary Fig. 4 online resource), and the carrier of an NF2 LoF GV had three syn- or metachronous tumors affecting the breast, uterus, and ovary (Fig. 3a, Supplementary Fig. 6 online resource).Fig. 3. Spectrum of personal syn-/metachronous non-brain tumors and familial tumors in glioma patients with pathogenic GVs. a Number of glioma patients with GVs in the specified genes and syn- or metachronous non-brain tumors at the specified sites. b, c Familial tumor spectrum of glioma patients with ATM GVs (b) or BRCA2 GVs (c), the most frequently affected CPGs in this study. Breast tumors were diagnosed in four first- or second-degree relatives of glioma patients with BRCA2 GVs (c). The pedigrees of glioma patients with GVs in other CPGs and suspected glioma risk genes are shown in Supplementary Figs. 1–7 online resource. It is not specified whether an individual is alive or deceased. BTF brain tumor family with one glioma patient and at least another brain tumor patient; CPG cancer predisposition gene; Dx age at diagnosis of primary tumor; GF glioma family with at least two glioma patients; GV germline variant; NA not available; No. number; NOS not otherwise specified; TF tumor family with one glioma patient and at least another tumor patient; V variant; WT wildtype; y years
The tumor spectrum in the families of glioma patients with pathogenic GVs in CPGs or suspected glioma risk genes is shown in three-generation pedigrees (also indicating the age at tumor diagnosis, Fig. 3b-c, Supplementary Figs. 1–7 online resource) and specified in Supplementary Table 2a online resource. In ATM GV carriers and their families, tumors of the brain (8/15 tumors, 53%), breast (2/15, 13%), neck, prostate, stomach, thyroid, and uterus (1/15, 7% each) were diagnosed (Fig. 3b). The ATM GV carrier and his brother in family Fam004 were diagnosed with glioblastoma at almost the same age (61 and 65 years, Fig. 3b). The median age at tumor diagnosis in all affected family members of ATM GV carriers with available data (n = 12) was 45 years. In BRCA2 GV carriers and their families, tumors of the brain (6/17, 35%), breast (4/17, 24%), skin (3/17, 18%), lung (2/17, 12%), bladder, and colon (1/17, 6% each) were observed (Fig. 3c). Breast tumors were diagnosed in first- or second-degree female or male relatives of glioma patients with BRCA2 GVs in 3/5 families, strongly suggesting that the breast tumor patients also carry the BRCA2 GV and that it is the tumor-predisposing variant in these families.
Characterization of tumors from patients with GVs in CPGs associated with DNA damage response
DNA damage response genes were affected by 22/77 (28.6%) of the different pathogenic GVs (Figs. 1b, 2a). Therefore, we determined the expression of markers of DNA double-strand breaks (DSB, phospho-H2AX), homology-directed repair of DSB (RAD51), and repair of DNA single-strand breaks (PARP1) as well as the expression of ATM and BRCA2 in available FFPE glioma sections from carriers of GVs in four genes conferring homologous recombination repair (HRR) deficiency, if mutated, i.e., ATM (n = 3 gliomas), BRCA2 (n = 3 gliomas), FANCA (n = 1 glioma), and SDHA (n = 2 gliomas) (Supplementary Fig. 8 online resource). A semi-quantitative analysis of ATM and BRCA2 staining revealed a trend towards a lower mean nuclear IRS for ATM in gliomas from ATM GV versus non-ATM GV carriers, and for BRCA2 in gliomas from BRCA2 GV versus non-BRCA2 GV carriers (Fig. 4a, b). The mean nuclear IRS of phospho-H2AX indicating DSB accumulation was lowest in gliomas from SDHA versus ATM, BRCA2, and FANCA GV carriers, although the differences were not statistically significant (Fig. 4c), possibly because SDHA variants are least directly linked to impaired DSB repair. Gliomas from SDHA GV carriers with less DSB accumulation showed a trend towards a higher mean nuclear IRS of RAD51 than those from ATM, BRCA2, and FANCA GV carriers with more DSBs (Fig. 4c, d). The mean nuclear IRS of PARP1 was significantly higher in gliomas from BRCA2 GV carriers than in those from ATM GV carriers (Fig. 4e).Fig. 4. Characterization of FFPE glioma sections from patients with GVs in the CPGs ATM, BRCA2, FANCA, and SDHA directly or indirectly associated with DNA damage response by immunohistochemistry and semi-quantitative analysis of the nuclear IRS. a, b The mean IRS of nuclear ATM (a) or BRCA2 (b) expression was lower in patients with versus without GVs in ATM or BRCA2, respectively. c–e To assess DNA damage and DNA damage response, the nuclear expression of the markers for DNA DSBs (phospho-H2AX, c), homology-directed repair of DSBs (RAD51, d), and repair of DNA single-strand breaks (PARP1, e) was determined. A4 astrocytoma, IDH-mutant, CNS WHO grade 4; CNS central nervous system; CPG cancer predisposition gene; DSB double-strand break; FFPE formalin-fixed paraffin-embedded; Gb4 glioblastoma, IDH-wildtype, CNS WHO grade 4; GV germline variant; IRS immunoreactivity score; pt patient; pts patients; WHO World Health Organization; w/o without
Other characteristics of primary or recurrent gliomas of 56 patients affected by at least one pathogenic GV in a CPG and/or a suspected glioma risk gene determined by approach 1 are listed in Supplementary Table 2b online resource. In gliomas (n = 4) from patients with MUTYH, PMS2, or POLE GVs, the median TMB was elevated (7.28, range: 6.02–8.59 mutations per megabase, Supplementary Table 2b online resource) compared to that previously defined for gliomas (2.6 mutations per megabase) [78].
Possible targeted therapy options for glioma patients with pathogenic GVs in certain CPGs
Most of the genes affected by GVs were CPGs (27/46, 58.7%, Fig. 1b, 2a). Pathogenic GVs in CPGs were detected in 48/213 (23%) glioma patients (Fig. 5a). In some tumor entities, it has been effective to target the molecular tumor phenotypes caused by 10 of the identified CPGs, if mutated, including HRR deficiency [43], high TMB [48], activation of the epidermal growth factor receptor (EGFR) [46], and p16^INK4A^ dysfunction/deficiency (preclinical evidence) [57]. As 24/48 (50%) glioma patients were affected by GVs in one of these 10 CPGs, they may be possible candidates for a molecularly targeted therapy, i.e., with PARP inhibitors (16/24, 67%), immune checkpoint inhibitors (ICI) (5/24, 21%), EGFR tyrosine kinase inhibitor (TKI) mono- or combination therapy (2/24, 8%, EGFR is expressed and Tyr1068-phosphorylated in two glioblastomas of patients with EGFR GVs, Supplementary Fig. 9 online resource), or CDK4/6 inhibitors (1/24, 4%) (Fig. 5b).Fig. 5. Additional treatment options may be effective in 11% (24/213) glioma patients with a familial and/or personal history of tumors. a The fraction of glioma patients with pathogenic GVs in CPGs is 23% (48/213). b For half (24/48) of the glioma patients with pathogenic GVs in CPGs, targeted treatment options exist that may potentially improve the outcome of patients with the respective GVs. ^#^, Loss-of-function variants in succinate dehydrogenase genes, including SDHA, lead to an accumulation of the oncometabolite succinate in the tumor that suppresses the homologous recombination DNA repair pathway [75]; ^*^, preclinical evidence only [57]; CPG cancer predisposition gene; GV germline variant; PARP poly(ADP-ribose) polymerase; TKI tyrosine kinase inhibitor
Discussion
In this study, we aimed to extend the knowledge on genes and variants impacting the genetic risk for glioma development by screening 213 adult glioma patients from 206 families with presumed tumor predisposition for germline variants by WES. With respect to CPGs, our findings strongly implicate BRCA2 pathogenic GVs in the tumorigenesis of adult gliomas (in line with [32, 51]), confirm that GVs in ATM, BRIP1, CDKN2A, MUTYH, PMS2, POLE, SDHA, TP53, and other CPGs are associated with glioma risk [15, 32, 51], and newly propose GVs in EGFR, GBA1, GJB2, and other CPGs as glioma predisposition candidates (Supplementary Tables 4, 5 online resource). GVs in CPGs were detected in 48 (23%) glioma patients with presumed tumor predisposition analyzed here, half of whom may be candidates for therapy options targeting the molecular tumor phenotype caused by their GVs (Fig. 5). Of the suspected glioma risk genes mutated in glioma patients here, two each were associated with cell adhesion, ion transport, vesicle transport, or immune response. Of the novel candidate genes mutated more frequently in the glioma than in the in-house control cohort, at least two had functions in metabolism or ion transport, respectively.
The BRCA2 gene, which encodes an essential homology-directed DNA DSB repair factor [56], was one of 10 affected CPGs involved in DNA damage response, making up 37% of all mutated CPGs, and was implicated in adult glioma risk using both approaches. Heterozygous pathogenic GVs in BRCA2 are associated with HRR deficiency and susceptibility to female breast, male breast, esophageal, gastric, pancreatic, ovarian, and prostate cancer [41, 55]. Similar to our findings, BRCA2 variants have been linked to pediatric brain tumors. In Fanconi anemia group D1 patients with biallelic pathogenic GVs in BRCA2, brain tumors, particularly medulloblastoma and astrocytoma, were found in the first decade of life in almost half (15/27) of the cases [2], and glioblastoma in childhood was also reported [21, 64]. In a meta-analysis, heterozygous ClinVar LP/P BRCA2 GVs were significantly more frequent in 876 children and adolescents with brain tumors than in a control cohort [38]. The frequency of heterozygous ClinVar pathogenic BRCA2 GVs in unselected adult glioma patients ranged from 3/764 (0.4%) [32] to 3/152 (2%) [51], compared to 5/206 (2.4%) glioma patients with presumed tumor predisposition analyzed here. All BRCA2 GVs identified here were LoF variants resulting in a reduced BRCA2 expression and an increased PARP1 expression, on average, in glioma sections compared to gliomas from non-BRCA2 GV carriers, thus impacting the molecular tumor phenotype.
The most frequently mutated gene, ATM, encoding a serine/threonine protein kinase, is also involved in DNA DSB repair, i.e., by phosphorylating proteins to initiate DNA damage response [40]. Biallelic ATM GVs cause ataxia telangiectasia characterized by a predisposition to tumors, including medulloblastoma [28] and cerebellar astrocytoma [53], immunodeficiency, and cerebellar degeneration among other features [68]. Heterozygous ATM GV carriers also have an increased cancer risk, particularly of female breast cancer [76]. Our data of heterozygous ATM GVs in 2.9% glioma patients with presumed tumor predisposition corroborate and extend the findings of ATM GVs in 1/152 (0.7%) glioma patients [51] and 3/304 (1.0%) glioma families [15]. In this study, heterozygous GVs in ATM predisposed mainly to IDH-mutant astrocytoma at a median age of 36 years, compared to 58 years at primary glioma diagnosis in patients without GVs, mainly affected by glioblastoma. The significantly younger age at glioma diagnosis of ATM GV carriers may at least partly be explained by the younger median age of patients presenting with IDH-mutant astrocytoma (38 years) [9], compared with a peak incidence of IDH-wildtype glioblastoma between 55 and 85 years [44]. Four of 6 ATM GVs detected in glioma patients here were LoF variants (two were ClinVar LP/P variants) that resulted in decreased ATM expression in the glioma tissue compared to gliomas from non-ATM GV carriers, thus impacting the molecular phenotype of the tumor. An effect of ATM variants on the radio-sensitivity of gliomas was previously reported, with significantly higher 1-year in-field control rates after radiation therapy in IDH-wildtype high-grade gliomas with versus without somatic pathogenic ATM variants [35].
Two genes associated with the repair of DNA replication errors were also recurrently affected, e.g., POLE, a DNA proofreading repair gene, and PMS2, a mismatch repair (MMR) gene. While heterozygous GVs in POLE and PMS2 cause polymerase proofreading-associated polyposis or hereditary non-polyposis colorectal cancer, respectively, predisposing to colorectal cancer [58, 59], they have also been linked to glioma risk [5, 15, 32, 34, 60, 80], in line with our data.
Around 20% of the mutated CPGs and 50% of the identified novel candidate genes of this study are functionally involved in metabolism, for instance of mitochondria (SDHA, TRMT5), lysosomes (GBA1), and peroxisomes (PHYH). SDHA encoding a subunit of the Krebs cycle enzyme succinate dehydrogenase is among the nuclear genes associated with the Leigh syndrome spectrum [50] that impact mitochondrial energy metabolism. Heterozygous SDHA LoF variants cause the pheochromocytoma/paraganglioma syndrome and lead to the accumulation of the oncometabolite succinate with various cellular consequences, e.g., on HRR [39, 75, 83]. Similar effects are seen due to elevated levels of the oncometabolite D-2-hydroxyglutarate in gliomas with activating variants of the IDH1 and IDH2 genes encoding the isocitrate dehydrogenase enzymes [19, 39]. In this study, two LoF variants (one of which was ClinVar LP/P) in SDHA possibly impacting succinate levels were detected in three patients with IDH-wildtype glioblastoma suggesting that the accumulation of succinate in glioblastomas with normal D-2-hydroxyglutarate levels may have similar oncogenic effects as the accumulation of D-2-hydroxyglutarate in IDH-mutant astrocytomas, and thus contribute to glioblastoma development. Similarly, glucosylceramide and glucosylsphingosine that are stored in the CNS, among other organs, in Gaucher disease type 2 and 3 caused by biallelic GVs in the GBA1 gene [73] may act as protumorigenic agents to increase the risk of glioblastoma and other tumors. This hypothesis would be in line with our finding of two different heterozygous ClinVar LP/P GVs in GBA1 in three glioblastoma patients, and an increased risk of hematological malignancies and solid tumors in patients with Gaucher disease [22].
Other gene functions implicated in glioma risk in this study include signal transduction (e.g. EGFR), cell adhesion (e.g. GJB2), immune response (e.g. IFIH1 and SAMHD1), and ion transport (e.g. CFTR). EGFR, encoding a transmembrane receptor tyrosine kinase acting in the PI3K/AKT/mTOR, MAPK, and other pathways, is among the genes most frequently activated by gene amplification, genetic rearrangements, and single nucleotide variants in IDH-wildtype glioblastomas [10]. While somatic EGFR variants in glioblastomas rarely affect the kinase domain [10], two of the three GVs in EGFR identified in the germline of glioma patients here and GVs in families with lung cancer affect the intracellular domain of EGFR [42]. The GJB2 gene encodes connexin 26, a gap junction protein associated with autosomal recessive and autosomal dominant non-syndromic deafness, and autosomal dominant hearing loss syndromes combined with skin disorders [62]. Interestingly, here we detected two ClinVar LP/P GJB2 GVs in three glioma patients, newly implicating pathogenic GVs in GJB2 in glioma risk. If biallelically mutated, IFIH1 and SAMHD1 are among the genes causing the autosomal recessive Aicardi-Goutières syndrome (AGS), a type I interferonopathy leading to an early-onset progressive encephalopathy with basal ganglia calcifications [18]. While one AGS patient with biallelic SAMHD1 GVs and chronic lymphocytic leukemia has been described [17], heterozygous GVs in SAMHD1 have been detected in a multiple myeloma family and significantly associated with prostate cancer risk [13, 52]. Here, we identified a heterozygous ClinVar LP/P SAMHD1 GV and a heterozygous IFIH1 GV with a CADD score > 30 in one patient each with an IDH-mutant glioma, providing further evidence for a link between AGS genes and glioma risk that we had previously described [6]. In that report, deleterious GVs in the AGS genes ADAR and RNASEH2B were detected in families and patients with astrocytomas or glioblastomas as well as with prostate cancer [6]. Biallelic GVs in the CFTR gene encoding an ATP-binding cassette transporter that functions as a chloride channel cause cystic fibrosis mainly affecting the lung, gastrointestinal tract, and skin [33, 65, 67]. Cases of pancreatic cancer and adenocarcinoma of the ileum have been reported, and a susceptibility for colorectal cancer has been observed in cystic fibrosis patients [7]. In addition, the prevalence of pathogenic CFTR GVs in the colorectal cancer population was significantly higher than expected suggesting an increased cancer risk not only in cystic fibrosis patients, but also in heterozygous CFTR GV carriers [7]. Similarly, we found heterozygous CFTR GVs to be significantly more frequent in glioma patients with presumed tumor predisposition compared to controls, suggesting a link to glioma risk.
In this study, 24/213 (11%) glioma patients with presumed tumor predisposition carried GVs in CPGs that potentially sensitize them to targeted therapies not routinely used in glioma patients, such as PARP, immune checkpoint, EGFR, or CDK4/6 inhibitors (Fig. 5). PARP inhibitors target DNA damage repair pathways leading to synthetic lethality of tumor cells with HRR deficiency, e.g., due to variants in genes such as ATM or BRCA2 [27]. Treatment with the PARP inhibitor olaparib provided a significant benefit over standard therapy in patients with a BRCA GV and metastatic breast or pancreatic cancer [26, 66], and a high response rate in patients with ATM GVs and metastatic prostate cancer [49]. Novel PARP inhibitors, such as AZD9574, are currently being developed that penetrate the blood–brain barrier and can be used in the treatment of brain tumors [72]. Blood–brain barrier penetrant PARP inhibitors may be effective in glioma patients carrying GVs in ATM, BRCA2, BRIP1, FANCA, and SDHA identified here that are associated with HRR deficiency, and may act synergistically with radio-, chemo-, and immunotherapy [70]. Advanced solid tumors with high TMB could have a robust response to ICI therapy [48]. Causes of high TMB include defects in MMR and polymerase proofreading repair due to pathogenic variants in PMS2 or POLE, respectively [11]. Therefore, five glioma patients identified here with heterozygous GVs in genes conferring a high TMB, i.e., PMS2, POLE, and MUTYH, may be candidates for treatment with ICIs, such as nivolumab or pembrolizumab, shown to be effective in individual glioblastoma patients carrying homozygous PMS2 GVs or a heterozygous POLE GV [8, 31], and in a pediatric trial of refractory malignancies with high TMB including glioblastoma patients with biallelic PMS2 GVs [20]. EGFR-TKIs, such as erlotinib, have shown high response rates in patients with non-small cell lung cancer and activating variants in the intracellular EGFR kinase domain [61], and may represent a potential therapeutic option for our patients carrying EGFR GVs with glioblastomas expressing Tyr1068-phosphorylated EGFR, which was associated with erlotinib sensitivity in preclinical lung cancer models [69]. Interestingly, erlotinib in combination with the chemotherapeutic agent gemcitabine showed efficacy in a patient with a pancreatic ductal adenocarcinoma carrying the EGFR:c.2189T>G p.(L730R) GV [54] that was also detected in a glioblastoma patient here. CDK4/6 inhibitors may be a future treatment modality for glioma patients with pathogenic CDKN2A variants causing melanoma-astrocytoma syndrome [77] detected here in one CDKN2A GV carrier with an anaplastic astrocytoma and a melanoma. According to preclinical data, CDKN2A deficiency sensitizes IDH-mutant glioma to CDK4/6 inhibitors [57]. As an increasing number of compounds is under preclinical investigation for molecular targeted therapy, additional treatment options for glioma patients with GVs in other genes may arise in the future.
In conclusion, 48/213 (23%) glioma patients with a presumed tumor predisposition carried at least one deleterious heterozygous GV in a CPG, identifying a hereditary syndrome linked to an increased risk of gliomas and other tumors in them and their families with implications for surveillance and, in around half of the cases, a potential for targeted treatment options. The genes implicated in glioma risk play roles in DNA damage response, e.g., ATM, BRCA2, PMS2, POLE, or diverse other processes including metabolism, signal transduction, and cell cycle regulation. Our data provide genes of interest for germline testing in glioma patients with a familial or personal medical history of tumors.
Supplementary Information
Below is the link to the electronic supplementary material.Supplementary file1 (PDF 3355 KB)