Phylogenetics within Camassia (Asparagaceae): examining difficult taxonomy and unusual variation using genomic restriction-site-associated DNA sequencing data
Jenny K. Archibald, Susan R. Kephart, Patrick J. Monnahan, Kathryn E. Theiss, Theresa M. Culley

TL;DR
This study uses genomic data to clarify the taxonomy and evolutionary history of Camassia plants in North America.
Contribution
The paper provides new phylogenetic insights into Camassia using RADseq data, revealing taxonomic ambiguities and evolutionary patterns.
Findings
Camassia species show cohesive phylogenetic groupings, but some subspecies are not monophyletic.
C. howellii and C. leichtlinii appear to have diverged early in the genus's evolution.
Geographic patterns and potential new species were identified within Camassia populations.
Abstract
Diversification of Camassia (Asparagaceae) in North America has shaped variety in morphological, ecological, and reproductive traits, and resulted in a classification with ambiguity in taxon boundaries, including numerous putative subspecies. Phylogenetic analyses of restriction-site-associated DNA sequences (RADseq) allowed new insights into its evolution and taxonomy, enhancing understanding of basal relationships, geographic patterns, taxonomic boundaries, and potential new species. A total of 157 individuals in 71 populations across all 15 putative taxa of Camassia and 42 outgroup individuals in 21 populations from Hastingsia and Chlorogalum were sampled and genomic libraries were generated using the modified single-digest RADseq method known as multiplexed shotgun genotyping. Assembly in ipyrad included targeted comparisons across a range of parameters that influence homology…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
 Figure 1
Figure 1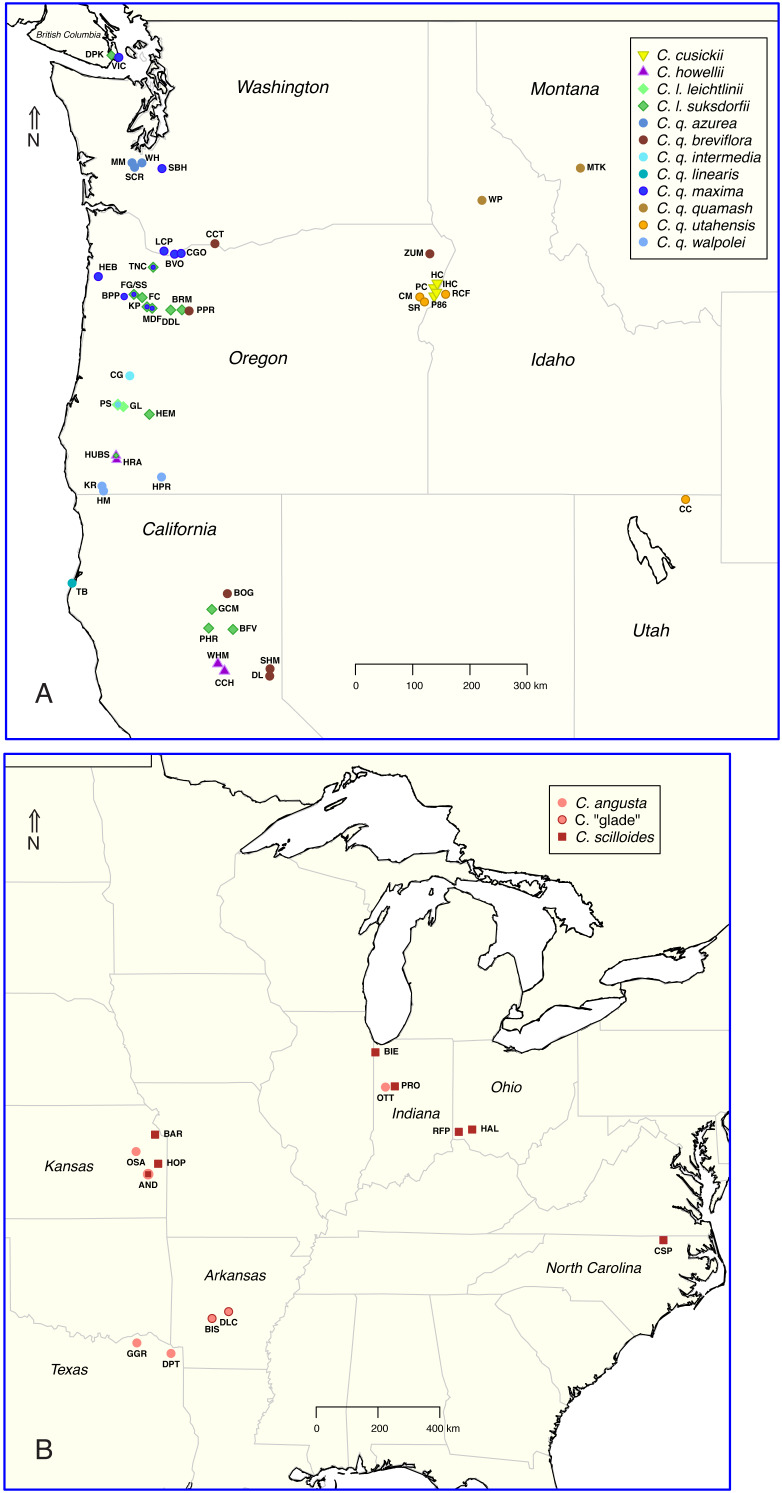 Figure 2
Figure 2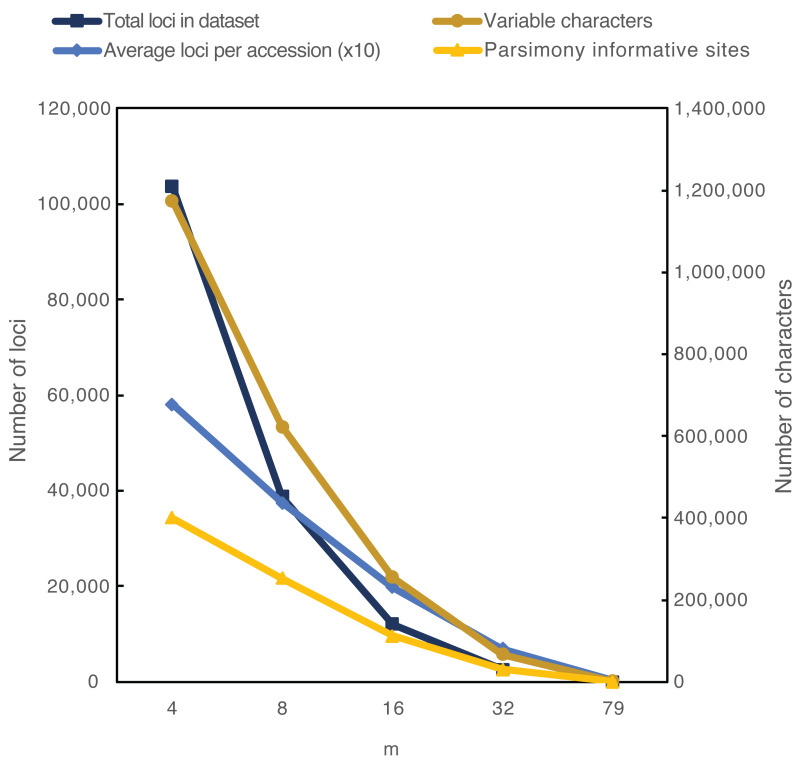 Figure 3
Figure 3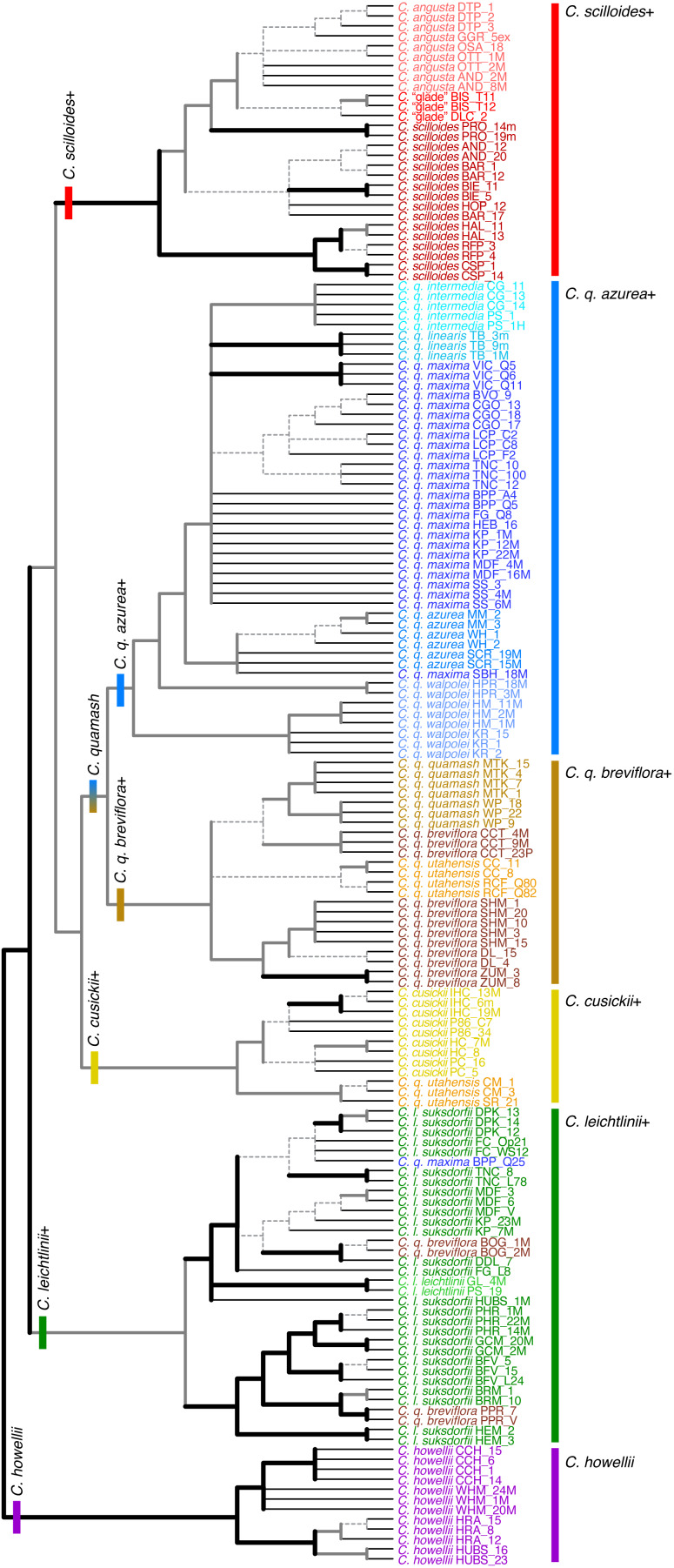 Figure 4
Figure 4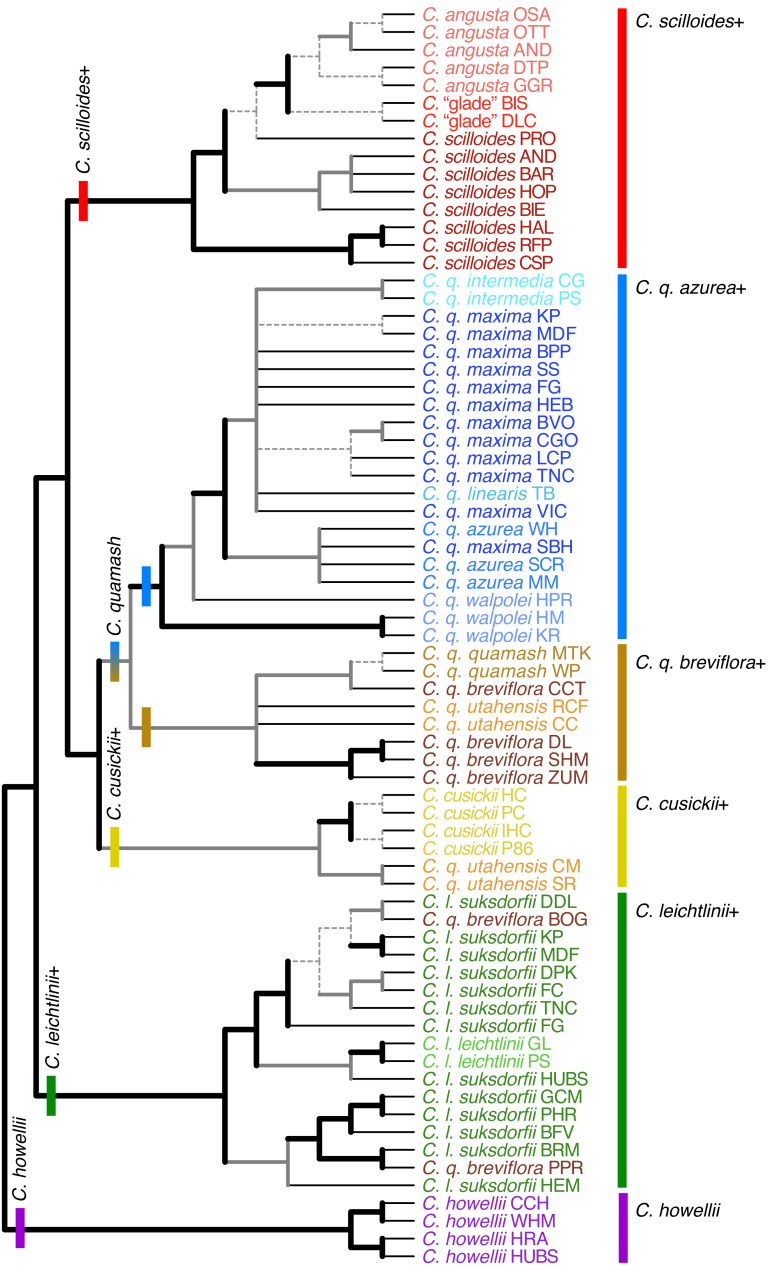 Figure 5
Figure 5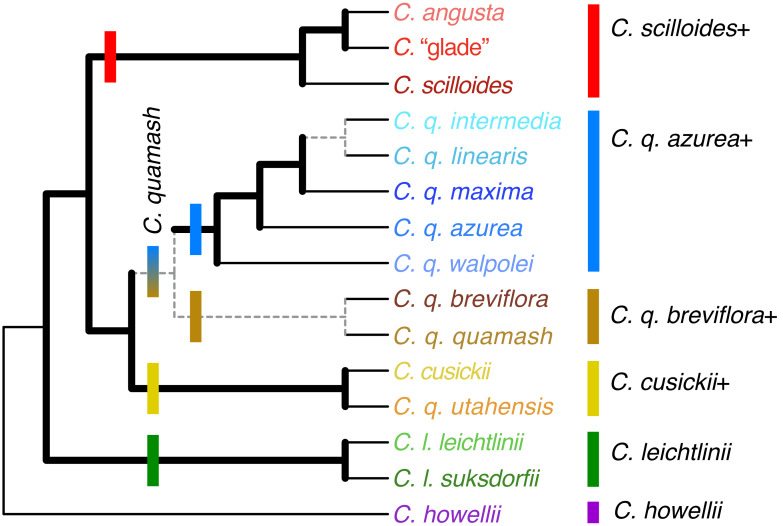 Figure 6
Figure 6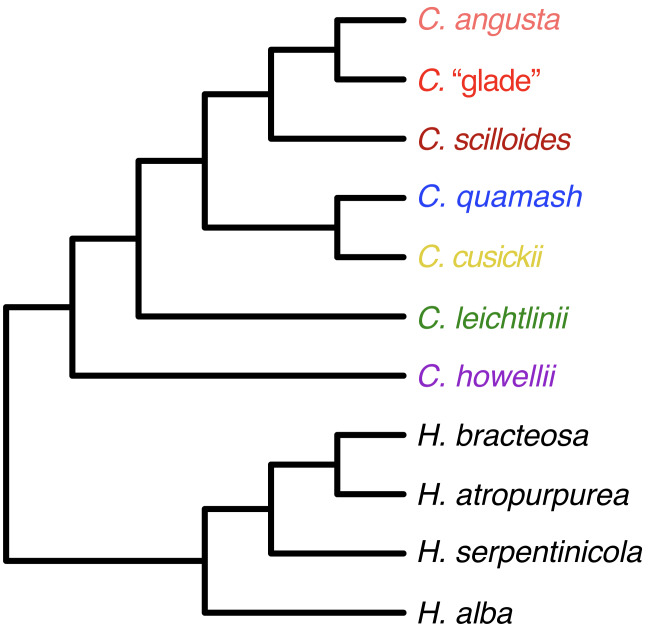 Figure 7
Figure 7- —NSF DEB 1145847 grant
- —NSF DBI 1262795 REU site
- —The Biodiversity Institute and the Department of Ecology and Evolutionary Biology at the University of Kansas (KU)
- —The Earthwatch Institute
- —M.J. Murdock Trust
- —Native Plant Society of Oregon
- —Willamette University
- —The Washington Native Plant Society
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsPlant and Fungal Species Descriptions · Plant Diversity and Evolution · Botany, Ecology, and Taxonomy Studies
Introduction
The extraordinary diversity of taxa and characters across the tree of life also reflects a diversity of mechanisms that allowed those lineages to diverge (De Queiroz, 2007). A thorough understanding of this diversity requires integration of phylogenetic, morphological, and ecological information, which is known as an integrative or iterative taxonomy approach to species delimitation (Dayrat, 2005; Padial & De La Riva, 2010; Yeates et al., 2011). As a case study for this type of approach, we are examining the plant genus Camassia Lindl. (Fig. 1; Asparagaceae; e.g., Kephart et al., 2025). Despite its tractable size of six named species, this genus encompasses diversity of various types, connected to a surprisingly complex taxonomy (Gould, 1942; Ranker & Hogan, 2002). Distributed within North America, some endemic species range across less than 100 km^2^, while others spread beyond 1,000 km^2^. The generic distribution encompasses a disjunction; four species are found in the Western USA and adjacent regions in Canada with a center of diversity in the Northwest US (C. cusickii S. Watson, C. howellii S. Watson, C. leichtlinii (Baker) S. Watson, and C. quamash (Pursh) Greene), while two species occur farther east: in the Midwest and southern US (C. angusta (Engelm. & A. Gray) Blank. and C. scilloides (Raf.) Cory; Ranker & Hogan, 2002). The two disjunct groups are separated by over 1,300 km at their closest point. One species (C. quamash) has eight subspecies (Gould, 1942), raising questions as to why such putative infraspecific diversity occurs without full separation into species and making application of taxon names difficult in the field. Morphological distinctions among subspecies and even among some species are not always clear (Kephart, 2015). Taxa in this genus also vary in ecological preferences (such as edaphic differences and prairie vs. wooded habitats; Merritt, 2021) and reproductive traits (such as in diurnal anthesis timing and flowering season; Archibald pers. obs., 2013; Ranker & Schnabel, 1986; Watson, 1889). There is hybridization between some infrageneric taxa and reproductive barriers between others (Uyeda & Kephart, 2006). Varying components (geography, morphology, ecology, and reproductive traits) allow examination of differing potential paths for diversification, likely having roles in the current levels of genetic isolation or the initial divergence of taxa (Coyne & Orr, 2004). A clearer understanding of taxonomy is also valuable for conservation efforts, such as in confirming that the locally appropriate taxon is used in restoration efforts (Kephart, 2015) and identifying distinct evolutionary lineages that may benefit from independent management plans. Many of these taxa sustain pollinator, herbivore, and human communities, historically and today (Carney et al., 2021; Parachnowitsch & Elle, 2005; Sultany, Kephart & Eilers, 2007).
Examples of morphology across Camassia.(A) C. leichtliniii ssp. leichtlinii population PS, (B) C. leichtlinii ssp. suksdorfii FC, (C) C. quamash ssp. walpolei KR, (D) C. quamash ssp. maxima MDF, (E) a prairie with individuals of C. scilloides BAR, and (F) C. angusta OSA. See Appendix I for an explanation of population codes. Photo credit: JK Archibald.
This paper provides a robust estimate of phylogeny as a key tool for further investigations of evolution within Camassia and for informing work to organize its diversity into a usable and meaningful classification. We also employed the phylogenetic results in combination with other knowledge on the biology of these organisms to examine specific unresolved taxonomic and phylogenetic questions in the genus, described below. Prior phylogenetic work on this group includes broader analyses in Agavoideae (Asparagaceae, APG, 2009; APG, 2016; Chase, Reveal & Fay, 2009). Analyses of one nuclear and two chloroplast loci by Archibald et al. (2015) and analyses of four chloroplast regions by Halpin & Fishbein (2013) strongly demonstrated the reciprocal monophyly and sister relationship of Camassia and Hastingsia S. Watson, with Chlorogalum Kunth s.s. (Taylor & Keil, 2018) sister to the Camassia–Hastingsia clade. Fishbein et al. (2010) provided the first phylogenetic trees across Camassia, sequencing two chloroplast regions and sampling at least one member of each species and subspecies, and the phylogenetic framework produced by Archibald et al. (2015) had particular focus on Camassia and its sister genus Hastingsia. The two studies supported different roots for Camassia, which could alter infrageneric appraisals of character evolution and biogeography. Although these prior studies provided a significant step forward in understanding evolutionary patterns within Camassia, many questions remain, from the basal rooting of the genus to relationships among and within species.
One species of interest is C. leichtlinii. Most treatments recognized it as a separate species (Gould, 1942; McNeal, 2012; Ranker & Hogan, 2002), and allozyme analyses suggested that C. leichtlinii and C. quamash largely remain genetically distinct. However, many sites show close sympatry between the two species, and some hybrid individuals have been identified (Uyeda & Kephart, 2006). Covering an extensive latitudinal range from southern Canada through California in the US, C. leichtlinii occurs both near and far from other species of Camassia. A more expansive study across populations of this widespread species would help clarify relationships with other western Camassia.
Moreover, relationships remain unclear among the eight subspecies of the other widespread western species, C. quamash. Both Fishbein et al. (2010) and Archibald et al. (2015) inferred two main clades of C. quamash individuals: a C. q. azurea+ clade comprised of C. quamash subspecies azurea (A. Heller) Gould, intermedia Gould, linearis Gould, maxima Gould, and walpolei (Piper) Gould, and a C. q. breviflora+ clade comprised of subspecies breviflora Gould, quamash, and utahensis Gould (Archibald et al., 2015). Yet, instead of being placed with other C. quamash in prior phylogenies, several populations of C. q. utahensis were placed with another species instead (C. cusickii). Overall, many relationships within these two clades were unresolved.
Difficult questions also remain in the more eastern C. scilloides complex, which may include two derivative species along with its namesake. There has been long-standing disagreement on whether C. angusta should be recognized as a separate species from C. scilloides (Gould, 1942; Steyermark, 1961). Ranker & Schnabel (1986) integrated studies of allozyme diversity, morphology, and phenology of this pair and concluded they were separate species. Those authors recommended a phylogenetic test of whether C. angusta is a derivative of C. scilloides, but sampling in prior phylogenetic studies was insufficient to address this question. Additionally and more recently, a new potential derivative taxon (referred to as C. “glade”) was discovered in Arkansas (Merritt, 2021), and it has not yet been included in a phylogenetic study.
Here we contribute the first in-depth phylogenetic analyses that include multiple samples and multiple populations from each putative infrageneric taxon, employing genome-level molecular markers. As a reduced-representation genomic sequencing approach, restriction-site-associated DNA sequencing (RADseq; Andolfatto et al., 2011; Baird et al., 2008) supplies many variable markers across the genome without requiring full genomic sequences, often allowing insights into even rapidly evolving clades, such as those within the Camassia species complexes. This method has previously proven useful for answering a range of biological questions in non-model species (Andrews et al., 2016). Its utility for inferring phylogenetic relationships among closely related taxa has been supported by multiple studies (Anderson et al., 2024; Cariou, Duret & Charlat, 2013; Chase, Stankowski & Streisfeld, 2017; Cruaud et al., 2014; Eaton & Ree, 2013; Frajman et al., 2019; Hipp et al., 2014; Leaché & Oaks, 2017; Marincek et al., 2024; Massatti, Reznicek & Knowles, 2016; Rubin, Ree & Moreau, 2012; Wagner et al., 2013). Although a loss of signal is seen with greater age, a simulation study by Rubin, Ree & Moreau (2012) supported utility of RADseq for phylogeny inference in clades as old as 40–60 myo. The Camassia clade is relatively young, estimated at approximately 5.22 million years old (myo) while a Camassia–Chlorogalum clade was estimated to be 6.85 myo (Smith et al., 2008; Hastingsia was not sampled).
In addition to providing a more robust phylogenetic framework across the genus through increased population and locus sampling, we specifically focused on these unresolved questions: (1) What are the basal relationships and root within Camassia? (2) In C. leichtlinii, do phylogenetic patterns vary with geography and are they affected by sympatry with congeners? (3) How do relationships within and among the many subspecies of C. quamash relate to their biology and taxonomy? (4) What is the status of C. quamash ssp. utahensis and C. cusickii in terms of their monophyly and potential sister relationship? and (5) For the more eastern taxa, we asked: (a) Where is the C. scilloides complex placed relative to the rest of Camassia, (b) is C. angusta supported as a derivative of C. scilloides, and (c) what is the placement of the potential new taxon C. “glade”?
Materials & Methods
Taxon sampling and DNA extraction
Sampling within Camassia comprised 157 individuals from 71 populations, with one to five plants per population (usually two to three) and all 15 putative taxa (Appendix I, Fig. 2). These included six named species, one with eight subspecies and one with two subspecies, and one unnamed potential segregate (Table 1). Population sites were indicated by alphanumeric codes, usually with two or three characters (Appendix I). Morphologically variable, widespread taxa were sampled in more depth, with emphasis on C. leichtlinii and C. quamash, especially C. quamash ssp. maxima. We included both allopatric and sympatric populations, when possible, for species with overlapping ranges. Species and subspecies were identified in the field using author expertise, especially S. Kephart who has published extensively on this genus, including floristic treatments (Kephart, 2015; Kephart, 2018). We also used local input (see Acknowledgements) and consulted prior research and taxonomic treatments (e.g., Gould, 1942; Ranker & Hogan, 2002; Ranker & Schnabel, 1986). Population boundaries were determined based on geographic distance combined with information on likely pollinator foraging distances and other barriers to gene flow (Cant et al., 2005; Danner et al., 2016). Outgroup sampling included some or all the populations collected for a separate phylogenetic study of Hastingsia (K Theiss et al., unpublished data, 2018) along with up to four populations from Chlorogalum (details below).
Maps showing the distribution of sampled populations of Camassia in North America, primarily in the (A) western and (B) midwestern to southern US.
Table 1: Taxa currently described or hypothesized within Camassia, with their sampling for this study and full known distribution (Kephart, Kephart & Robinson, 2019; Kephart, 2015; Kephart, 2018; Ranker & Hogan, 2002).
Leaf material (∼20 mg or more per individual) was dried on silica gel from each population. DNA was extracted from leaf samples using standard hexadecyltrimethylammonium bromide (CTAB) methods (Doyle & Doyle, 1987) or a DNeasy Plant Mini Kit (Qiagen, Germantown, Maryland, USA). Quantity and quality of DNA were assessed using QuBit (2.0 fluorometer, Invitrogen, Waltham, Massachusetts, USA) and Nanodrop (ND-1000 Spectrophotometer, ThermoScientific, Waltham, Massachusetts, USA), respectively. Samples were chosen based on a target of 260/280 ratios between 0.9 and 2.5 and 260/230 ratios between 0.5 and 3.
DNA sequencing
Genomic libraries were generated for 96 samples at a time, using a modified single-digest RADseq method known as multiplexed shotgun genotyping (Andolfatto et al., 2011; Baird et al., 2008). Samples were diluted to 5 ng/µL and 10 µL of template DNA was aliquoted to each well (50 ng total). A few samples with lower concentrations were diluted to 2.5 ng/µL and included in multiple wells to maximize coverage. DNA from each individual was digested with restriction enzyme AseI (NEB Biolabs, Ipswich, Massachusetts, USA), and then unique 6-bp bar-coded adapters were ligated to each sample (T4 DNA ligase, Enzymatics, Germantown, Maryland, USA). The samples were pooled and purified in separate steps using isopropanol precipitation and AMPure bead purification (Agencourt, Beckman Coulter, Brea, California, USA). Afterwards, the library was size-selected, targeting fragments of 275 bp using a Pippin Prep (Sage Science, Beverly, Massachusetts, USA), followed by another round of bead purification. Polymerase chain reaction (PCR) (14 cycles) was conducted using Phusion High-Fidelity PCR Master Mix (NEB Biolabs, Ipswich, Massachusetts, USA) and indexed primers that bind to common regions in the adaptors. PCR-products were again purified with AMPure beads, using a 0.8 × bead/template volume ratio to remove small (primer-dimer) fragments. Sequences were generated in three Illumina (San Diego, California, USA) HiSeq 2500 PE100 lanes at the Genome Sequencing Core, University of Kansas, Lawrence, Kansas, USA. Each lane typically contained up to 96 pooled individuals, including some that were sequenced for separate studies. A 10% phiX spike-in was included for all lanes to provide additional sequence complexity.
Molecular data processing and phylogenetic analyses
We demultiplexed the FastQ files into sample-specific sequence files using custom scripts developed by John K. Kelly (Appendix S1; see Supplemental Data with this article), and processed the demultiplexed files using pyRAD v. 3.0.5 through ipyrad v. 0.7.30, using the latter for the final analyses (Eaton & Overcast, 2020). For the final sets of analyses, we ran a de novo assembly of the RADseq reads in ipyrad under the “pairgbs” datatype. We increased quality filtering stringency using filter_adapters=2 and phred_Qscore_offset=43 and employed a range of parameter settings to assess the influence on results, with particular emphasis on the influence of clust_threshold (here referred to as “c”) and min_samples_locus (here referred to as “m”). The c parameter indicates the threshold of similarity required for two loci to be identified as homologous and clustered together, and it was set at 70, 80, 90, or 95 for the final analyses. The m parameter indicates the minimum number of accessions that must have sequence data at a locus for that locus to be retained in the dataset, and it was set at 4, 8, 16, 32, or 79. For analyses of the full Camassia dataset (157 accessions), those m values correspond to retaining a locus found in 2.5%, 5%, 10%, 20%, and 50% of the accessions, respectively. The analysis with m79 resulted in very poorly supported trees and so was not explored further. These and a series of preliminary analyses supported c90, m04 as producing the most informative dataset in this genus and in the Camassia–Hastingsia clade, and so those settings were used for the rooting analyses. Due to a major observed increase in missing data when including the outgroups (see Results), we used unrooted analyses of *Camassia-*only datasets to infer relationships within the genus, while determining likely roots using multi-genera analyses. After the aligned output files were produced, we manually removed accessions with fewer than 100 loci from the datasets for all subsequent analyses.
We inferred phylogenetic relationships on concatenated alignments using maximum likelihood (ML) with the General Time Reversible (GTR)+ Γ model of nucleotide substitution in RAxML version 8.1.20–8.2.11 (Stamatakis, 2014); rapid bootstrapping analyses with 100 replicates provided relative clade support. To better account for potential gene-tree heterogeneity, we also estimated coalescent-based trees using singular value decomposition quartets (SVDquartets) (Chifman & Kubatko, 2014) as implemented in PAUP* v. 4a157 through 4a165 (Swofford, 2003). This method allows a coalescent-based approach, while being computationally feasible with datasets of these sizes and without estimating gene trees. Gene trees can often be unreliable when based on RADseq loci, which are relatively short. SVDquartets has also been shown to be valid with some gene flow (Long & Kubatko, 2018), which is likely to have occurred across some taxonomic boundaries in this genus (Uyeda & Kephart, 2006). We exhaustively sampled all quartets and assessed support using 100 bootstrap replicates. Preliminary analyses with 5,000 bootstrap replicates did not notably change the inferred support. We conducted individual, population, subspecies, and species tree inference under the multispecies coalescent (MSC) model. Using these two inference methods (concatenated ML and SVDquartets) allowed us to look at the combined signal of the many, relatively short RADseq loci, while also better accounting for possible deep coalescence and other factors that may cause conflict between gene trees. We also used full loci rather than Single Nucleotide Polymorphisms (SNPs), with paired-end sequencing and 100 bp reads to obtain as much information as possible per locus. Preliminary field observations suggested that C. “glade” may be a separate taxon. We treated it as a “species” for the purpose of the species-based tree, due to uncertainty in its status. However, if we had merged it with C. angusta or set it as a subspecies of C. angusta, its inferred placement would not have changed (see Results).
Initial analyses were run with and without one individual (C. quamash ssp. maxima_BPP_Q25). Its morphology suggested influence of gene flow from plants of C. leichtlinii at the same site and its inferred phylogenetic placement was far from the other individuals sampled from its population and even from other C. quamash (see Results and Discussion). The inclusion of this individual did not affect other inferred relationships in the individual-based phylogenetic analyses, but it did influence the population-based analyses using SVDquartets. So, we excluded C. q. maxima_BPP_Q25 for those analyses and most rooting analyses, leaving 156 accessions of Camassia.
As prior datasets have disagreed on the root of Camassia (Archibald et al., 2015; Fishbein et al., 2010), and given the major potential impact of outgroup choice on inferred roots (De la Torre-Bárcena et al., 2009; DeSalle, Narechania & Tessler, 2023), we compared several different rooting analyses. We also focused on reducing missing data, as there is a known tendency for increased missing data at higher phylogenetic distance with RADseq datasets (Eaton et al., 2017). The different rooting datasets used were: (1) the complete final set of all sampled accessions of Camassia, Hastingsia, and Chlorogalum, (2) all sampled accessions of Camassia (except C. q. maxima_BPP_Q25, discussed above) and the 20 accessions of Hastingsia that each shared ≥100 loci with each of ≥10 accessions of Camassia, (3) the accessions from Camassia and Hastingsia that each shared ≥100 loci with ≥1 accession from the other genus, and (4) all Camassia and Hastingsia accessions that each had ≥2,000 total sequenced loci (Table 2). Custom scripts developed by John K. Kelly (University of Kansas) counted the number of shared and matching loci for each pair of accessions. One additional rooting dataset included only four accessions of Hastingsia, focusing on those that shared a greater number of loci (≥500) with accessions of Camassia. However, results from those analyses were highly incongruent with all others, including nesting one or more individuals of Hastingsia deep in the Camassia clade. A close examination of those results suggested it was a spurious result of missing data, and so they will not be discussed further.
Table 2: Characteristics of RADseq datasets of Camassia and outgroups from Hastingsia and Chlorogalum.
Results
RADseq loci across and within the genera, and rooting of Camassia
The average sequencing depth for loci from each accession ranged from 10 to 117 across Camassia, Hastingsia, and Chlorogalum, while the average number of usable reads per accession was 21,212 (reads_consens in ipyrad). Within the focal 157 accessions of Camassia, the overall average depth was 33 and that did not change with varying ipyrad parameters. The number of loci within the Camassia datasets varied from over 137,433 to only 47, dropping with higher m values (Table 2, Fig. 3). When we used m32 as our largest m value (excluding m79), the smallest number of total loci was 2,550. The average number of loci per individual was much lower than the total number of loci across individuals, demonstrating the lack of overlap of many loci across individuals and a correspondingly high level of missing data. The number of parsimony informative sites showed a similar trend, ranging from over 400,398 to 631 depending on the parameters (with a minimum of 30,722 if excluding the m79 analyses).
Comparisons of datasets for 157 accessions of Camassia with ipyrad parameters of c90 and the indicated values of m.The m parameter indicates the minimum number of accessions that must have sequence data at a locus for that locus to be retained in the dataset. The average numbers of loci per accession have each been multiplied by 10 to allow clearer visualization.
Decreasing missing data by excluding more loci resulted in lower clade support. With the m parameter at 04 (including a locus in the dataset even if only 2.5% of the accessions had that locus), ML and SVD analyses resolved 100 and 58 clades with >90% bootstrap (BSt) within Camassia, respectively. In contrast, analyses of the same accessions with m32 (requiring 20.4% accessions) resolved less than half that number of strongly supported clades (46 and 26 clades). Less taxonomic cohesion also was evident at higher m values. For example, nine putative taxa were resolved as monophyletic with SVD analyses of the m04 individual dataset but only five with m32. At the population level, there was a 38% and 18% decrease in strongly supported monophyletic populations for ML and SVD analyses, respectively, when changing from m04 to m32. The effect of sacrificing signal to avoid missing data was even more clear if we required loci to be found in at least 50% of the accessions (m79 for the Camassia dataset); only 47 loci remained in that dataset and there were almost no strongly supported clades in the resulting trees.
Many more loci were shared within Camassia than between it and the outgroup genera. On average, pairs of accessions of Camassia shared 252 loci (based on 12,246 pairs of accessions), whereas Camassia–Hastingsia pairs shared 53 loci (5,652 pairs). Regardless, all relationships among the genera had bootstrap support of 100%. The overall levels of support and relationships inferred within Camassia were similar when loci were processed and analyzed separately for this genus or alongside those for the other two genera, despite the steep increase in missing data. That consistency was seen for both deep and shallow relationships within the genus. One well-supported exception was the ML placement of the C. cusickii+ clade, where the ML analyses of datasets with multiple genera moved the C. cusickii+ clade into the C. quamash clade, sister to the C. q. breviflora+ clade (BSt = 100%).
The basal relationships in Camassia were inferred as one of two possibilities by rooting analyses. Either C. howellii–leichtlinii was sister to the rest of Camassia, or C. howellii and C. leichtlinii formed a grade at the base of Camassia, with C. howellii sister to the rest of Camassia. Maximum likelihood analyses supported the former, whereas SVD analyses supported the latter. We chose the C. howellii root when illustrating relationships (Figs. 4–7), given that this root was supported at all taxonomic grouping levels and by taxonomically broader analyses using data from nuclear and chloroplast loci (Archibald et al., 2015). Not illustrated, but possibly of interest to some readers are the relationships among our few sampled Chlorogalum accessions: two populations of C. pomeridianum (DC.) Kunth var. pomeridianum formed a strongly supported clade sister to C. p. var. minus Hoover (with variable support), and this clade was sister to two individuals from a population of C. p. var. divaricatum (Lindl.) Hoover. This differed from Archibald et al. (2015), which instead supported C. p. var. pomeridianum as sister to C. p. var. divaricatum.
Individual-level phylogenetic relationships inferred using RADseq data from Camassia.Relationships are summarized for 10 analyses conducted in both RAxML and SVDquartets with these ipyrad settings: c80m04, c90m04, c90m08, c90m16, c95m04. Regardless of parameter settings (with few exceptions), thick black branches were resolved and strongly supported (>90% Bootstrap, BSt)a, and thick gray branches were resolved but with variable levels of supportb. Dashed branches varied in resolution and support but show the relationships inferred and most strongly supported by the majority of analyses. Inferred relationships were collapsed into polytomies if there was no dominant pattern supported across analyses with different parameters. Thin black branches lead to tips. Major clades are indicated with bars at their base and to the right of taxon names. These analyses were unrooted; the tree shows one of two possible roots supported by separate rooting analyses. The alternative resulted in a C. howellii–C. leichtlinii+ clade as sister to the rest of Camassia. See text for details. aIf a relationship had >90% BSt for all analyses except for one with >75% BSt, the line was left as thick black. bIf a relationship was resolved by all analyses except for one, and the conflicting relationship had <50% BSt support, the line was left as thick gray.
Relationships within Camassia
Consistency across parameter sets
Representing analyses run using different parameter sets, different levels of grouping, and both ML and SVD optimization, our tree figures summarize phylogenetic relationships inferred across 25 different analyses of the Camassia-only datasets (Figs. 4–7). Our tree of species-level relationships also summarizes four of the rooting analyses of Camassia and Hastingsia (Fig. 7). Overall, the deeper relationships in Camassia were fully resolved and well supported, whereas some relationships within species were not strongly supported.
Analyses using RAxML and SVD were largely consistent; the most common difference was that some relationships were not as strongly supported in SVD analyses as in RAxML. All inferred relationships were consistent across SVD analyses with different grouping of individuals (i.e., without grouping, grouping as populations, as subspecies, and as species). In this case, “consistent” means that most inferred clades were identical, with occasional minor differences where a relationship was partially supported by one set of analyses but unresolved by another. For example, partial support was found in cases where some analyses in a set resolved a given relationship, and others did not (indicated by dashed branches in the trees). In other cases, the relationship was inferred by all analyses in the set but with variable support (indicated by solid gray branches in the trees). However, even the relationships indicated by dashed branches were often identically resolved by the individual, population, subspecies, and species analyses. Some populations or taxa were not completely monophyletic in individual-based analyses. When forced to be monophyletic by grouped SVD analyses, the placement of those lineages was unsurprising, following the majority of their accessions. Specific cases are discussed below.
General patterns of relationships
Each of the species of Camassia was supported as fully monophyletic, monophyletic excepting a few outliers, or paraphyletic. Camassia howellii, C. cusickii, and C. angusta s.l. (i.e., if encompassing C. “glade”) were each monophyletic in all analyses. Camassia scilloides formed a grade at the base of a strongly supported clade of C. angusta and C. “glade”. The 79 individuals sampled from 34 populations of the 8 subspecies of C. quamash formed a largely monophyletic group, except for a few populations that moved out and disrupted the otherwise monophyletic C. leichtlinii (Figs. 4 and 5).
Population-level phylogenetic relationships inferred using RADseq data from Camassia.Relationships are summarized for five analyses conducted in SVDquartets with these ipyrad settings: c80m04, c90m04, c90m08, c90m16, c95m04. Regardless of parameter settings (without exceptions), thick black branches were resolved and strongly supported (>90% bootstrap, BSt), and thick gray branches were resolved but with variable levels of support. Dashed branches varied in resolution and support but show the relationships inferred and most strongly supported by the majority of analyses. Inferred relationships were collapsed into polytomies if there was no dominant pattern supported across analyses with different parameters. Thin black branches lead to tips. Major clades are indicated with bars at their base and to the right of taxon names. These analyses were unrooted; all separate population-level rooting analyses supported the root shown in the figure.
Within C. leichtlinii, C. l. leichtlinii was monophyletic but nested within clades of C. l. suksdorfii (Greenm.) Gould. Even with broad sampling of 29 accessions from 15 populations of this species, many relationships were strongly and consistently supported across all analyses. The species was divided into two strongly supported clades and many populations were supported as monophyletic (none were contradicted, but some were unresolved). Relationships within C. leichtlinii were identical when comparing the individual-level and population-level phylogenies, except that the C. l. leichtlinii clade and C. l. suksdorfii_HUBS formed a polytomy with the rest of the members of one of the two subclades of C. leichtlinii in individual-level analyses (Fig. 4) and were a well-supported sister clade to the rest of that subclade in population-level analyses (Fig. 5). The C. leichtlinii+ clade included phylogenetic outliers from three populations of C. quamash: two populations of C. q. breviflora (BOG and PPR) and one individual of C. q. maxima (population BPP).
The main C. quamash clade was divided into two well-supported clades: the C. q. azurea+ clade and the C. q. breviflora+ clade (Figs. 4–6). Each subspecies of C. quamash was restricted to one of these two main clades, with a few exceptions (see below). The C. q. azurea+ clade included a well-supported subclade of C. q. intermedia, C. q. linearis, and C. q. maxima, sister to C. q. azurea, with C. q. walpolei forming a grade at the base. Two narrowly distributed taxa were each well supported as monophyletic: C. q. intermedia (two populations, five individuals sampled) and C. q. linearis (one population, three individuals). In contrast, relationships among the 27 individuals (12 populations) of wide-ranging C. q. maxima were generally not well resolved beyond forming a clade with C. q. intermedia and C. q. linearis, with two outliers. One population of C. q. maxima (SBH, with one individual sampled) was placed in the otherwise-monophyletic C. q. azurea clade rather than with other members of its subspecies. The other outlier (C. q. maxima _BPP_Q25) fell farther, in the C. leichtlinii clade. However, other sequenced individuals from this population were placed with the main group of C. q. maxima individuals. As noted in the Methods, C. q. maxima_BPP_Q25 was removed from some analyses due to possible influence of gene flow. Forming a grade at the base of the C. q. azurea+ clade, two populations of C. q. walpolei (HM, KR) formed a clade, whereas population HPR was sister to a large clade containing the other subspecies of the C. q. azurea+ clade.
Subspecies-level phylogenetic relationships inferred using RADseq data from Camassia.Relationships are summarized for five analyses conducted in SVDquartets with these ipyrad settings: c70m04, c90m04, c90m16, c90m32, c95m04. Regardless of parameter settings (without exceptions), thick black branches were consistently resolved and strongly supported (>90% bootstrap, BSt); all those branches had bootstrap values of 100%, excepting a few from the c70m04 analysis. Dashed branches varied in resolution and support but show the relationships inferred and most strongly supported by the majority of analyses. Thin black branches lead to tips. Major clades are indicated with bars at their base and to the right of taxon names. These analyses were unrooted; all separate subspecies-level rooting analyses supported the root shown in the figure.
Species tree relationships inferred using RADseq data from Camassia and Hastingsia.Relationships are summarized for five analyses conducted in SVDquartets of Camassia accessions alone (using ipyrad parameters c70m04, c90m04, c90m16, c90m32, and c95m04) and four of Camassia and Hastingsia (using c90m04). All relationships had 100% bootstrap support in all analyses, except for the H. bracteosa–H. atropurpurea clade in one analysis (97% BSt) and the sister relationship between C. howellii and the rest of Camassia in another (99% BSt).
The C. q. breviflora+ clade included C. q. breviflora, C. q. quamash, and two of four populations of C. q. utahensis. The monophyly of C. q. quamash had mixed support, with stronger support from individual-based analyses than some population-based analyses. None of the analyses resulted in a monophyletic C. q. breviflora. One population of C. q. breviflora (CCT) was instead usually inferred as sister to C. q. quamash, although the support for this division was weak. Two populations of C. q. breviflora (BOG and PPR, two individuals sampled for each) were each monophyletic and strongly supported as members of different subclades within the C. leichtlinii+ clade in both the individual and population-based trees. When C. q. breviflora was forced to be monophyletic by subspecies-level SVD analyses, the lineage remained with the rest of C. quamash. Finally, C. q. utahensis was divided by individual- and population-level analyses, with two populations (CC and RCF) in the C. q. breviflora+ clade and two populations (CM and SR) placed sister to C. cusickii. This C. cusickii–C. q. utahensis_CM,SR clade was then sister to the entire C. quamash clade. In the five subspecies-level SVD analyses, C. q. utahensis was forced to be monophyletic and was sister to C. cusickii (BSt ≥ 94% for all) rather than with the remainder of its species, but there was either reduced support for the monophyly of the remainder of C. quamash (three analyses: c90m04, c90m16, c90m32) or that monophyly was disrupted by the movement of the C. cusickii–C. q. utahensis clade to be nested with other members of the C. q. breviflora+ clade (two analyses: c70m04 and c95m04, also found in some rooting analyses, as noted above). In both cases, forcing monophyly of C. q. utahensis lowered support for some containing clades of that subspecies compared to analyses where populations were able to move separately within the phylogeny.
The more eastern clade of Camassia had the most notable difference between RAxML and SVD individual-based analyses. Some SVD analyses intermixed populations of C. “glade” and C. angusta, whereas those two groups were strongly supported as sister and reciprocally monophyletic by RAxML. Regardless, together C. angusta and C. “glade” formed a well-supported clade (Figs. 4–7).
Discussion
RADseq utility and missing data
We found general utility of RADseq for inferring relationships across recently diverged genera, species, and subspecies in this group, although not all relationships within subspecies were clear. These datasets were comprised of over 137,000 loci within Camassia, with over 400,000 parsimony informative sites. As seen elsewhere, the parameters used for filtering and determining homology of loci greatly influenced their numbers (Leaché et al., 2015; Takahashi, Nagata & Sota, 2014; Wessinger et al., 2016), and signal remained even with a large proportion of missing data (Crotti et al., 2019; Huang & Knowles, 2016; Lee et al., 2018). Fewer loci in RADseq datasets with higher m values was expected, although the loss of signal was more severe here than in some studies. For example, several studies generally focused on clades ranging in age from 2 to 16 myo (Appelhans et al., 2018; Maier et al., 2019; Sharples & Manzitto-Tripp, 2024) successfully used m values that included a locus if 13–60% of the accessions had sequences for that locus (Maier et al., 2019; Paetzold et al., 2019; Raheem et al., 2023; Sharples & Tripp, 2019). Comparing to Camassia at approximately 5.22 myo (Smith et al., 2008), setting m to require even as much as 20% sampling resulted in lowered support for many clades when compared to analyses with more loci (and more missing data). Insufficient sequencing effort can be a source of missing data in RADseq datasets (Eaton et al., 2017), but that was likely not a problem here, given our high sequencing depth (Andrews et al., 2016). We also saw decreases in the number of supported monophyletic taxa and populations when excluding loci to lower the amount of missing data. A taxon or population being non-monophyletic could accurately reflect the biology of that group. However, multiple identifiable taxa or populations being resolved as monophyletic in one set of analyses and not monophyletic in another set suggests loss of signal in the latter. The pull of C. cusickii+ towards the C. q. breviflora+ clade, seen primarily in some three-genus analyses, highlights one potential impact of missing data. This alternate placement may relate to an affinity between a member of the latter clade (C. q. utahensis) and C. cusickii (discussed below), and it was also inferred by two subspecies-level analyses where C. q. utahensis was forced to remain monophyletic. Regardless, and despite high levels of missing data, our datasets allowed resolution of consistent and well-supported phylogenetic hypotheses across a range of parameters for many relationships (Figs. 4–7).
The root and early diversification within Camassia
The monophyly of Camassia was very strongly supported across all multi-genera analyses (100% BSt). Our analyses highlighted the importance of careful rooting. Different analyses strongly supported different roots despite choosing outgroups from the closest relatives of this genus and choosing accessions to maximize signal. We found support for two likely roots for Camassia, either a C. howellii–C. leichtlinii+ clade (supported by ML analyses) or C. howellii alone, with C. leichtlinii+ then being sister to the remainder of Camassia (supported by SVD analyses at all grouping levels). In prior analyses using Bayesian and maximum parsimony, Fishbein et al. (2010) weakly supported C. howellii–C. leichtlinii+ as sister to the rest of Camassia using two chloroplast loci, while the combined ITS and cpDNA analyses of Archibald et al. (2015) found strong support for C. howellii as sister. Our data strongly reinforce those two hypotheses. In either scenario, we saw that the C. howellii and C. leichtlinii+ clades split early from the remainder of Camassia and the unrooted basal relationships within Camassia were identical and strongly supported across most analyses. Consistent with these conclusions, Gould (1942) proposed an early origin of both C. howellii and C. leichtlinii, especially C. howellii. This was based on its morphological similarity to related genera, particularly Chlorogalum. In the next section, we focus within C. leichtlinii, while Kephart et al. (2025) separately discuss integrative studies of C. howellii and its relationships with sympatric species, using morphology, microsatellite, and cpDNA data.
Geographic patterns and gene flow in the well-resolved and widespread Camassia leichtlinii
Camassia leichtlinii extends from the northern half of California in the US, northward into British Columbia in Canada and consists of two subspecies, of which, C. l. leichtlinii is native to just one county in Oregon (Fig. 2; Ranker & Hogan, 2002). In Oregon and northward, C. l. suksdorfii occurs from the coast to the slopes of the Cascade mountains, but rarely east of the Cascades (Fig. 1 in Gould, 1942), and it is often sympatric with C. quamash. In California, C. l. suksdorfii populations grow on both sides of the Cascades and in the Sierra Nevada mountains, and it has not been found in sympatry with C. quamash (data provided by the participants of the Consortium of California Herbaria, ucjeps.berkeley.edu/consortium/).
The subspecies differ in flower color, creamy-white in C. l. lecihtlinii and variable blue to deep violet, rarely white, in C. l. suksdorfii (Figs. 1A, 1B; Gould, 1942). Large, diurnal flowers, five to nine tepal veins, and oblong fruits separate C. leichtlinii from its potential sister species C. howellii, which has smaller, vespertine flowers, three to five tepal veins, and subglobose fruits (Kephart, 2015). Comparing western species in the genus, flowers of C. howellii and C. leichtlinii abscise at the pedicel tip if no fruit is matured, while C. cusickii and C. quamash retain spent flowers on intact pedicels (Kephart, 2015). Although C. leichtlinii is distinctive in morphology, habitat differentiation is less obvious. This species, like C. quamash, inhabits varied wet meadow, riparian, and oak savannah communities in diverse soil types from near sea-level to 2,400 m (Ranker & Hogan, 2002; Sultany, Kephart & Eilers, 2007). However, when in sympatry with C. quamash and C. howellii, C. leichtlinii prevails in partial shade or wetter microhabitats of open sites, rather than the more open areas where the other two species are found (Sultany, Kephart & Eilers, 2007).
The monophyly of C. leichtlinii was well-supported across our analyses (Figs. 4–6), with the addition of a few outliers from C. quamash, discussed below. This species had the strongest internal support across the phylogenies, with two main clades (Figs. 4 and 5). One clade included relatively high elevation populations (ranging from around 1,100 to 1,670 m) and the other held low elevation populations (from around 50 to 460 m). This division is largely consistent with relationships inferred previously (Archibald et al., 2015; Fishbein et al., 2010), although two populations that were sampled by both studies (HUBS and BFV) were placed differently by analyses of Fishbein et al. (2010), C. l. suksdorfii_BFV was sister to the rest of C. leichtlinii+ and C. l. suksdorfii_HUBS was nested within C. howellii.
Considering our two clades within C. leichtlinii+, the montane (high-elevation) clade included five populations of C. l. suksdorfii, along with outlier C. q. breviflora_PPR. Relationships within this clade mirrored geography. A basal grade of Oregon Cascade populations (C. l. suksdorfii_HEM, BRM, and C. q. breviflora_PPR) fell with a nested subclade of three California populations (a subclade of C. l. suksdorfii_GCM–PHR from the Cascades, sister to BFV from the Sierra Nevadas). Our clade of low-elevation populations included 10 populations from both subspecies of C. leichtlinii and two populations of C. quamash. All 12 occur below 460 m, excepting one of the two outlier populations (C. q. breviflora_BOG at 1,889 m). Within this clade, some relationships reflected geographic patterns and others did not. The C. l. leichtlinii clade and C. l. suksdorfii_HUBS have a potentially close relationship (Fig. 5) and are also only ∼80 km apart geographically, compared to ∼170 to 690 km distance between the C. l. leichtlinii clade and any other population in this subclade. These three sites also lie along a well-traveled route today and historically for indigenous tribes and European settlers (Reinhardt, 2020), providing a strong possibility of human-mediated migration (Auffret, Berg & Cousins, 2014; Silcock et al., 2024). In contrast, a lack of geographic correspondence was seen with C. l. suksdorfii_DDL, which lies just 25 km from C. l. suksdorfii_BRM, but was placed in a separate subclade.
Given recognized hybridization with C. quamash (Uyeda & Kephart, 2006), it may not be surprising that a few individuals of putative C. quamash were resolved phylogenetically within the C. leichtlinii+ clade. However, we also saw support for the general integrity of these species’ boundaries, such as at the sympatric site PS, where sequenced individuals of C. l. leichtlinii and C. q. intermedia were each placed with their respective species in the phylogeny (Fig. 4). Overall, the C. leichtlinii+ clade is no more closely related to the C. quamash clade than it is to any other species of Camassia (Figs. 4–7). Considering current geography and opportunity for gene flow, outliers were strongly supported as members of both major subclades of C. leichtlinii, but all populations of C. leichtlinii in the high-elevation clade are allopatric with other species of Camassia, while most in the low-elevation subclade are sympatric with or grow within 2–5 km of C. q. maxima. As noted above, placement of outlier C. q. maxima_BPP_Q25 is most plausibly due to local introgression; other sampled individuals from this population lie within the C. quamash clade with their subspecies. At this site, C. quamash typically flowers 2–3 weeks before C. l. suksdorfii, limiting hybridization (Uyeda & Kephart, 2006). However, some likely hybrids have been collected, and this individual (Q25) was noted as having morphologically intermediate traits while appearing closer to the known morphology of C. q. maxima (S Kephart, pers. obs., 2010). The outlier individuals of C. q. breviflora are from populations BOG and PPR. Accession V of C. q. breviflora_PPR was also sampled by Fishbein et al. (2010) and resolved within a C. leichtlinii+ clade using chloroplast loci; its placement was ascribed to ancestral polymorphism or prior gene flow. Those authors excluded recent chloroplast capture, largely because the outlier fell in a subclade of geographically distant populations of C. leichtlinii. However, our results show C. q. breviflora_PPR as sister to a population not sampled by Fishbein et al. (2010), C. l. suksdorfii_BRM, which occurs within ∼4 km of this outlier. A separate, integrative study on Camassia populations in the California Floristic Province includes C. q. breviflora_BOG and allows a closer look at the morphology and phenology of this population compared to C. leichtlinii (Kephart et al., 2025). Similarly, population-level studies of C. q. breviflora_PPR, with sampling of additional individuals from this population and nearby populations of C. leichtlinii would be useful. This would be an additional check against potential misidentification or contamination of samples–although the likelihood of that occurring is low given careful specimen handling and detailed morphological examination to identify individuals. Regardless, deeper sampling would allow a more detailed view of genetic and morphological diversity, within and between the two species.
The many subspecies of C. quamash and a possibly porous species boundary with C. cusickii
The taxonomically and morphologically diverse C. quamash was subdivided into two well-supported clades (Figs. 4–6). Members of the C. q. azurea+ clade occur west of the Cascade mountains in the Pacific Northwest region of the United States, with a moderate climate and abundant rainfall; members of the C. q. breviflora+ clade occur in the rain shadow east of the Cascades within dry semi-arid and high desert areas with diurnal and seasonal temperature extremes (Fig. 2; Ranker & Hogan, 2002; Taylor & Hannah, 1999). We will discuss each subspecies within these two clades, with hypotheses for unexpected phylogenetic relationships, how they may connect to geographic patterns, and their taxonomic implications. Morphological traits have clearly diverged across these subspecies, but not always with sharp or universal distinctions among them.
Rethinking the subspecies of C. quamash west of the Cascades (the C. q. azurea+ clade)
Despite field, herbarium, and phylogenetic studies (Archibald et al., 2015; Fishbein et al., 2010; Gould, 1942), taxon boundaries have remained uncertain among the five westernmost subspecies of C. quamash, including C. q. walpolei, C. q. azurea, C. q. intermedia, C. q. linearis, and C. q. maxima. Possibly the most morphologically distinctive is C. q. walpolei, a subspecies known from southwest Oregon in the Siskiyou-Klamath region, growing in wet meadows or forest openings (Kephart, 2015). In his monograph of Camassia, Gould (1942) referred to C. q. walpolei as “the best defined subspecies of C. quamash”. It is morphologically distinct from others in the C. q. azurea+ clade in its primarily radial rather than zygomorphic symmetry, smaller flowers, and fruits with shorter pedicels and fewer seeds (Ranker & Hogan, 2002). This subspecies also differs from others in this clade in that ∼74% of sampled plants have tepals with three veins instead of five to nine (Gould, 1942; Kephart, 2015). The only subclade of C. q. azurea+ that was well supported by prior analyses comprised two populations of C. q. walpolei (Archibald et al., 2015); our increased population sampling revealed paraphyly of this subspecies, which formed a grade at the base of the C. q. azurea+ clade (Figs. 4 and 5). Although morphological traits can vary developmentally or among populations within subspecies of C. quamash (Kephart, 2015), C. q. walpolei is distinctive in its morphology and phylogenetic relationships, suggesting that it be maintained taxonomically.
Like C. q. walpolei, C. q. azurea has a limited range, but it grows on grassy mounds or in prairies of western Washington. Most prevalent in glacial outwash soils of counties near South Puget Sound and the Olympic Peninsula, C. q. azurea ranges as far north as Whidbey Island (Kephart, 2018). Only C. q. maxima comes close geographically to C. q. azurea, but C. q. maxima grows in wet meadows to rocky bluffs at unglaciated sites that are largely allopatric to C. q. azurea (Gould, 1942). Morphologically, several traits distinguish C. q. azurea from the other subspecies. In Gould’s key, the pale blue-violet perianths of C. q. azurea differ from the deeper blue-violet of C. q. maxima, although he notes intergradation in floral color and bulb traits of these subspecies in Washington. Spreading vs. appressed fruit pedicels separates C. q. azurea (spreading in ≥ 95% of individuals) from six of the remaining seven subspecies (fruits appressed to stems in ≥ 99% of individuals), while C. q. maxima is variable (60% of fruits appressed; Gould, 1942). Initial morphometric data reveal potential differences between C. q. azurea and C. q. maxima in bract length relative to pedicel length, but also variable floral color in C. q. maxima; thus, wider sampling is essential (Theiss & Kephart, 2015). An ecological difference between C. q. azurea and C. q. maxima is seen in attacks of cecidomyiid flies (Dasineura camassiae) that induce camas to form flower galls, with putative host specificity (Barosh & Kephart, pers. obs., 2012; Gagné, Barosh & Kephart, 2014). Galls occurred in over half of populations of C. q. azurea that we observed but have not been found in any populations of C. q. maxima (N >18), even at sites that house other gall-forming Camassia taxa (Barosh & Kephart, pers. obs., 2012).
Our RADseq analyses provided the first phylogenetic evidence for a potentially monophyletic C. q. azurea (Figs. 4 and 5); it was previously within a polytomy (Archibald et al., 2015; Fishbein et al., 2010). Only our single individual from C. q. maxima_SBH falls within the otherwise-monophyletic C. q. azurea. Due to geographic proximity, prior gene flow from C. q. azurea to C. q. maxima_SBH is plausible via pollen transport by bees or seed dispersal from floristically similar glacial outwash prairies in Thurston County (Kruckeberg, 1991). For example, glacial meltwater may have connected C. q. azurea_WH and C. q. maxima_SBH (separated by ∼32 km). It is yet unknown whether these two subspecies can cross-fertilize. In all, while questions remain about these variable taxa, our data support some distinctiveness of C. q. azurea, thus we do not recommend taxonomic changes with this subspecies at present.
The remaining subspecies in the C. q. azurea+ clade formed a well-supported subclade in the RADseq phylogenies, comprising C. q. intermedia, C. q. linearis, and C. q. maxima (Figs. 4–6). The ranges of C. q. intermedia and C. q. linearis are relatively narrow and fully disjunct from each other, in southwestern Oregon and northwestern California, respectively (Ranker & Hogan, 2002). Despite its limited range in California, C. q. linearis occurs in diverse sites, representing north coastal bluffs, wetlands, and montane slopes of the coast range (K Theiss, pers. obs., 2024; Gould, 1942). In contrast, strictly inland C. q. intermedia grows in open or wooded wet areas (Kephart, 2015). Broader-ranging C. q. maxima, in Oregon, Washington, and British Columbia, is also disjunct from C. q. linearis, but overlaps with C. q. intermedia in Lane County, Oregon (Gould, 1942). Within C. quamash, these three subspecies are the most difficult to separate using morphology. Leaves of C. q. maxima tend to be slightly glaucous, while leaves of the other two subspecies are not glaucous (Gould, 1942; Ranker & Hogan, 2002). Connivent tepal withering at senescence distinguishes C. q. linearis from all other taxa in the C. q. azurea+ clade, whose tepals wither separately (Gould, 1942). Relatively pale flowers have been used to distinguish C. q. intermedia from C. q. maxima and C. q. linearis, but floral color is variable for C. q. maxima (noted above). Overall, the morphology of C. q. intermedia falls within the known ranges of C. q. maxima.
Earlier phylogenies had no well-supported resolution within this subclade (Archibald et al., 2015; Fishbein et al., 2010). Our analyses did not strongly infer relationships among most populations of C. q. maxima, but they did resolve a strongly supported C. q. linearis clade and a well-supported C. q. intermedia clade, both in a polytomy with C. q. maxima. The chloroplast tree of Fishbein et al. (2010) also supported the monophyly of C. q. linearis, but with just 0.67 posterior probability, while C. q. intermedia was weakly supported as not monophyletic.
Given its greater phylogenetic and morphological distinctiveness, and its geographic isolation, we support maintaining C. q. linearis as a subspecies, pending further study of this diverse subclade. Due to the noted lack of a clear morphological division between C. q. intermedia and C. q. maxima, and weaker phylogenetic evidence separating these subspecies, we propose this nomenclatural synonymy.
Camassia quamash (Pursh) Greene subsp. maxima Gould. Amer. Midl. Nat. 1942. 28:732–733.
Camassia quamash (Pursh) Greene subsp. intermedia Gould. Amer. Midl. Nat. 1942. 28:734–735.
The subspecies of C. quamash east of the Cascades (the C. q. breviflora+ clade) and a link to C. cusickii
As noted above, the second major clade of C. quamash included C. q. breviflora, C. q. quamash, and C. q. utahensis (Figs. 4 and 5). Farthest west from the other two and with the largest latitudinal range is C. q. breviflora, distributed from Washington to California. East of that range is C. q. quamash, distributed north of C. q. utahensis (Ranker & Hogan, 2002). Two of the sampled populations of C. q. utahensis fell within this C. q. breviflora+ clade (C. q. utahensis_CC, RCF), but two others were sister to C. cusickii (C. q. utahensis_CM, SR), separate from the rest of their species. Also occurring east of the Cascades, C. cusickii was once considered endemic to Oregon but additionally occurs across the border in Idaho (Adams County) and has disjunct populations in Washington (Kephart, Kephart & Robinson, 2019; Kephart, 2018).
Several morphological traits distinguish C. quamash and C. cusickii. Bulbs of C. cusickii are ellipsoid and clustered, while those of C. quamash are globose and rarely clustered. There are also rarely fewer than 10 leaves for C. cusickii and usually fewer than 10 for C. quamash (Kephart, 2018; Ranker & Hogan, 2002). Jewell (1978) noted differences in seed shape and indumentum, and that C. cusickii often has a densely flowered raceme, whereas C. quamash is more loosely flowered. A specific difference with C. q. utahensis may be tepal withering, which is often separate in C. cusickii and connivent in C. q. utahensis (Ranker & Hogan, 2002). Comparing morphology across the three subspecies of C. quamash in the C. q. breviflora+ clade, connivent tepals are also found in C. q. breviflora, but not in C. q. quamash, while a larger plant stature (20–70 cm) and distinctly bilateral flowers link C. q. utahensis and C. q. quamash, compared to the shorter C. q. breviflora (10–50 cm) with variable floral symmetry. For the latter two traits, C. cusickii has larger plants (50–90 cm) and variable floral symmetry (Kephart, 2015).
Jewell (1978) conducted a morphological, ecological, and flavonoid comparison of C. cusickii and C. quamash. He recommended that the two be maintained as separate species but noted intermediate specimens in the area of our C. q. utahensis_SR population, which fell in a clade sister to C. cusickii in our phylogenies. Jewell (1978) stated “Whereas these plants remain referrable to C. quamash var. utahensis in all the keys, they are intermediate in phenotypic expression between C. cusickii and C. quamash var. utahensis. This may presumably result from past hybridization and subsequent introgression”. The C. q. utahensis_CM population, which also clustered with C. cusickii in our and other analyses (Archibald et al., 2015; Fishbein et al., 2010), is only ∼10 km from SR.
Looking across our sampled populations, three of C. q. utahensis are within 15–23 km of our cluster of sampled populations of C. cusickii (Fig. 2), including the two that grouped with C. cusickii in the phylogeny (CM and SR), and one that did not (RCF; Figs. 4 and 5). The other population of C. q. utahensis (population CC) in the C. quamash clade is ∼520 km from this cluster of populations. Prior phylogenetic analyses differed in population sampling for C. q. utahensis and DNA regions used, with differences in some details of their inferences (Archibald et al., 2015; Fishbein et al., 2010). In all cases, some populations of C. q. utahensis group with C. cusickii, and others group with C. quamash near to C. q. breviflora and C. q. quamash. Fishbein et al. (2010) sampled three populations of C. q. utahensis that are not included in this study. Those populations were over 100–200 km from our C. cusickii sites, both east and west, and each grouped with C. quamash. In all, many populations of these two species were geographically close enough for potential gene flow, although a well-defined geographic pattern did not emerge for populations of C. q. utahensis that grouped with C. quamash versus those that grouped with C. cusickii.
Considering the monophyly of taxa across these clades, both prior phylogenetic studies sampled the same two populations of C. cusickii, which were not monophyletic in both cases (Archibald et al., 2015; Fishbein et al., 2010). We added two populations and multiple individuals from each population and resolved a well-supported C. cusickii clade (Figs. 4 and 5). When forced to be monophyletic by subspecies-grouped SVD analyses, C. q. utahensis was sister to C. cusickii (Fig. 5). In regard to the other two taxa in the C. q. breviflora+ clade, the monophyly of C. q. quamash was supported in these and prior analyses (Archibald et al., 2015; Fishbein et al., 2010), and each included the type locality (WP). The monophyly of C. q. breviflora was not fully supported due to a potential link between population CCT and the C. q. quamash clade, and due to outliers discussed above in the C. leichtlinii+ clade (Figs. 4 and 5).
Population genetic and morphological work are in progress to resolve this difficult complex (S Mortimer et al., unpublished data, 2025). Further study is needed, particularly with C. q. utahensis to verify if the phylogenetic division of its populations reflects gene flow across species and to address its lack of cohesion.
A progenitor-derivative complex: the C. scilloides+ clade
The three potential taxa of Camassia that occur in the Midwest and farther south in the US (Fig. 2; Merritt, 2021; Ranker & Hogan, 2002) formed a strongly supported clade (C. scilloides+) that was sister to the C. quamash–C. cusickii clade (Figs. 4–6). This result was consistent across ML and SVD analyses, but it differs from previous inferences. The chloroplast trees of Fishbein et al. (2010) and Archibald et al. (2015) strongly inferred the C. scilloides+ clade as sister to a C. q. breviflora+ clade, nested within C. quamash. Separate ITS analyses of Archibald et al. (2015) instead placed C. scilloides+ as sister to all remaining Camassia, except C. howellii, but with variable support (Jackknife = 56, BI posterior probability = 0.92). Combined ITS and chloroplast analyses in Archibald et al. (2015) were consistent with the chloroplast resolution. Geographically, the C. q. breviflora+ clade does contain the easternmost subspecies of C. quamash, but it is still separated by over 1,300 km from the more eastern C. scilloides complex (Ranker & Hogan, 2002). The pattern of relationships supported by our study is surprising in that it differs from both previous hypotheses, but it concurs with the taxonomy in keeping a largely monophyletic C. quamash.
The C. scilloides complex was initially recognized as just one species. Gould (1942) stated in his treatment of the genus that C. scilloides and C. angusta were “certainly not specifically distinct”, but that they might be recognizable as subspecies. Two later studies of this pair instead argued for recognition of C. angusta, based on differences in morphology, flowering season, and allozyme markers (Ranker & Schnabel, 1986; Steyermark, 1961), and both species were recognized in the Flora of North America (Ranker & Hogan, 2002). Ranker & Schnabel (1986) studied 9–11 populations of each species and found that they had “taxon-specific patterns of morphological and isozymic variation”, but had diverged little. The species differed statistically for six of nine morphological traits (despite considerable overlap). Phenological isolation was strong in sympatry: flowering in C. angusta begins 2–3 weeks after ending in C. scilloides, and this difference was generally maintained in common garden experiments. The geographic range and allozyme markers of C. angusta were largely subsets of those for C. scilloides. Ranker & Schnabel (1986) thus proposed these two species as a recent progenitor-derivative species pair. Our phylogenetic results supported this hypothesis, nesting a C. angusta–C. “glade” clade within lineages of C. scilloides. Our results also suggested genetic isolation between C. scilloides and C. angusta. We sampled five individuals from two sites where those species are sympatric; in all cases individuals were placed with members of their own species in the phylogeny (Figs. 4 and 5; C. scilloides and C. angusta from site AND, and C. angusta from site GGR; C. scilloides_GGR was not available for this study).
Proposed recently is C. “glade” (Merritt, 2021), a potential taxon that shares some traits with C. angusta and others with C. scilloides, along with its own unique features. For example, capsules are ovoid-ellipsoid in C. “glade” and C. angusta; whereas C. scilloides has subglobose capsules. The former two species also flower later in the spring compared to C. scilloides and both have tepals that become strongly connivent over the ovary and then dehisce following anthesis (Merritt, 2021). Like C. scilloides, C. “glade” has fewer sterile bracts and flowers relative to C. angusta, and its racemes are more tapered at the top. Variation in perianth color may be taxon specific, but further work is needed. The major distinction for C. “glade” is that it is endemic to glade habitats composed of edaphic grasslands with thin soils amid exposed bedrock outcrops, in contrast to the prairie, wooded, or riparian habitats of C. angusta and C. scilloides. The C. “glade” sites are all in Arkansas and include flat, seasonally wet shale glades, and seasonally wet rocky riverscour glades composed of shale and sandstone in the Ouachita Mountains and in Saline County (T Witsell, pers. comm., 2012; Merritt, 2021). Most of our analyses resolved C. “glade” as monophyletic and sister to C. angusta (Figs. 4 and 5), although some SVD analyses showed potential intermixing of populations of C. angusta and C. “glade”. The connection between the two was well supported by these data, as was their separation from C. scilloides, but without strong evidence for or against recognizing C. “glade” as a formal taxon separate from C. angusta. An ecological niche modeling study (B Merritt et al., unpublished data, 2025) and a microsatellite study (T Culley et al., unpublished data, 2024) of C. scilloides, C. angusta, and C. “glade” may support further consideration of C. “glade” taxonomically. A valuable next step would be to pursue a “common garden” experiment with the three purported taxa, to see if any of the distinctive morphological features of C. “glade” are phenotypic responses to its unique environment.
Conclusions
Overall, these RADseq analyses provided a robust framework for continued study of morphology, population genetics, ecology, and reproductive factors in Camassia. We found strong support of many relationships from the population to species levels, more so at deeper levels of the phylogeny. Comparing results with use of varying parameters for homology assessment and allowance of missing data allowed us to emphasize conclusions that are robust to changes in parameter decisions. The high level of missing data was overwhelmed in many cases by phylogenetic signal to allow inference of relationships. Basally, both C. howellii and C. leichtlinii separated early from the rest of Camassia. We inferred two possible relationships between the two species, with support for either a sister relationship or a grade, depending on the optimality criterion used. Within the widespread C. leichtlinii, relationships among many populations were strongly supported and patterns of diversification for some subclades suggested a geographic component in their diversification. The eight subspecies of C. quamash largely formed a monophyletic group, although a few outliers fell with C. leichtlinii or C. cusickii. In particular, C. q. utahensis was confirmed as being divided into two clades on the trees, one within C. quamash and the other sister to C. cusickii, which was itself supported for the first time as being monophyletic. Further investigation is needed to determine the status of C. q. utahensis. Relationships among other subspecies of C. quamash were not fully resolved but often showed some support for subspecies recognition. However, we recommend synonymizing C. q. intermedia with C. q. maxima. Although it may be monophyletic, C. q. intermedia was encompassed within C. q. maxima both morphologically and phylogenetically. The disjunct complex of species farther to the east in North America was strongly resolved as a monophyletic C. scilloides+ clade sister to the C. quamash–C. cusickii clade. Camassia angusta was supported as a derivative of progenitor C. scilloides, while a potential new taxon and edaphic specialist (C. “glade”) appeared either sister to or intermixed with C. angusta. Overall, the genus includes examples of clear-cut discrete species, a putative unnamed taxon, and potential admixture across current taxonomic boundaries, even involving currently allopatric populations.
Supplemental Information
10.7717/peerj.20438/supp-1Supplemental Information 1Scripts used to demultiplex FastQ files
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Anderson BM Binks RM Byrne M Davis R Hislop M Rye BL 2024 Revised taxonomy for two species complexes of Western Australian Isopogon (Proteaceae) using RA Dseq Taxon 7316118910.1002/tax.13129 · doi ↗
- 2Andolfatto P Davison D Erezyilmaz D Hu TT Mast J Sunayama-Morita T Stern DL 2011 Multiplexed shotgun genotyping for rapid and efficient genetic mapping Genome Research 2161061710.1101/gr.115402.11021233398 PMC 3065708 · doi ↗ · pubmed ↗
- 3Andrews KR Good JM Miller MR Luikart G Hohenlohe PA 2016 Harnessing the power of RA Dseq for ecological and evolutionary genomics Nature Reviews Genetics 17819210.1038/nrg.2015.28PMC 482302126729255 · doi ↗ · pubmed ↗
- 4APG III 2009 An update of the Angiosperm Phylogeny Group classification for the orders and families of flowering plants: APG III Botanical Journal of the Linnean Society 16110512110.1111/j.1095-8339.2009.00996.x · doi ↗
- 5APG IV 2016 An update of the Angiosperm Phylogeny Group classification for the orders and families of flowering plants: APG IV Botanical Journal of the Linnean Society 18112010.1111/boj.12385 · doi ↗
- 6Appelhans MS Reichelt N Groppo M Paetzold C Wen J 2018 Phylogeny and biogeography of the pantropical genus Zanthoxylum and its closest relatives in the proto-Rutaceae group (Rutaceae)Molecular Phylogenetics and Evolution 126314410.1016/j.ympev.2018.04.01329653175 · doi ↗ · pubmed ↗
- 7Archibald JK Kephart SR Theiss KE Petrosky AL Culley TM 2015 Multilocus phylogenetic inference in subfamily Chlorogaloideae and related genera of Agavaceae—informing questions in taxonomy at multiple ranks Molecular Phylogenetics and Evolution 8426628310.1016/j.ympev.2014.12.01425585154 · doi ↗ · pubmed ↗
- 8Auffret AG Berg J Cousins SAO 2014 The geography of human-mediated dispersal Diversity and Distributions 201450145610.1111/ddi.12251 · doi ↗