Metagenomic analysis of bacterial and viral communities of Aedes aegypti and Aedes albopictus
Shahzadi Asia Nadeem, Ijaz Ali, Hazrat Hussain, Ihsan Ullah, Wajid Ali, Khalid J. Alzahrani, Hamid Ali, Zarak Imtiaz Khan, Kasim Sakran Abass, Rafi ur Rahman

TL;DR
This study explores the bacterial and viral communities in Aedes aegypti and Aedes albopictus mosquitoes from Pakistan to better understand their role in arbovirus transmission.
Contribution
The study provides a metagenomic analysis of bacterial and viral communities in Aedes mosquitoes from specific regions in Pakistan.
Findings
Klebsiella pneumoniae and Acinetobacter baylyi were the most abundant bacteria in Ae. aegypti.
Pseudomonas putida and Brevundimonas diminuta were the most abundant bacteria in Ae. albopictus.
Siphoviridae viruses, including Escherichia phage HK639 and Lactobacillus phage 2, were prevalent in both species.
Abstract
The complicated relationship between the Aedes mosquito microbiome, arbovirus transmission and essential physiological processes, is extremely important. Microbial community plays a vital role in shaping vector biology, impacting critical aspects such as parasite replication within the vector, vector longevity, and ultimately, vector competence. Understanding the composition and function of the Aedes microbiome is therefore crucial for developing novel strategies to control arboviral diseases. Therefore, we aimed to identify prevalent bacterial and viral communities in Aedes mosquitoes from Pakistan. Ae. aegypti and Ae. albopictus were collected and from three different regions of Khyber Pakhtoonkhwa, Punjab and federal capital Islamabad. We isolated DNA and sequenced two pools of each species and conducted metagenomic analysis, identifying a variety of bacteria and viruses. We found…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
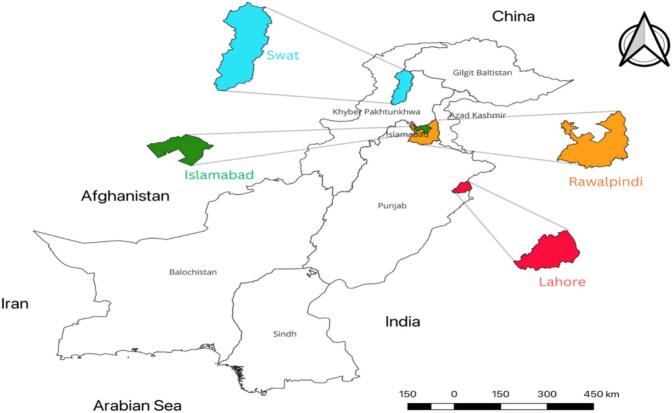 Figure 1
Figure 1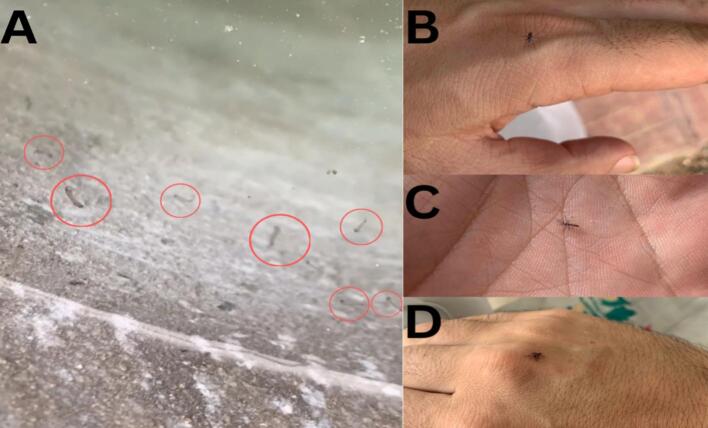 Figure 2
Figure 2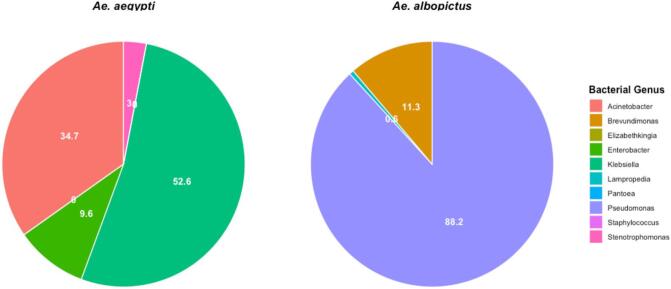 Figure 3
Figure 3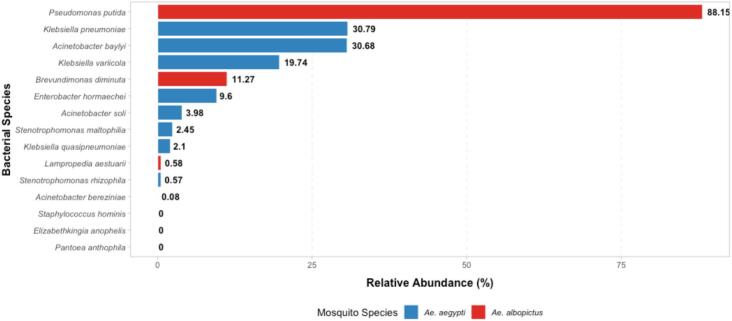 Figure 4
Figure 4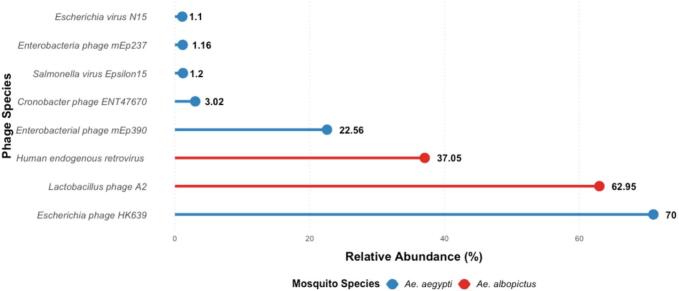 Figure 5
Figure 5Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsInsect symbiosis and bacterial influences · Mosquito-borne diseases and control · Studies on Chitinases and Chitosanases
Introduction
1
Vector-borne diseases pose a significant burden on public health and economic growth of a country. Specifically, arboviral diseases are one of the main concerns due to the rapid increase in the incidence and vast geographical distribution. Chikungunya virus (CHIKV), Zika virus (ZIKV) and dengue virus (DENV) are some of the arboviruses of medical importance which need active surveillance and control1, 2, 3, 4. In humans, CHIKV, DENV, and ZIKV are transmitted through the bite of Aedes aegypti and Aedes albopictus mosquitoes3, 4, 5. In Pakistan, dengue virus has established endemicity, expanding to previously unaffected regions. In federal capital Islamabad, confirmed dengue cases increased from 2438 in 2023 to 3970 in 2024 with 15 deaths (Personal communication). This substantial increase in dengue cases, coupled with high mortality highlights a rapidly escalating trend of dengue fever severity within the region.
Ae. aegypti is widely distributed all over the world, particularly in tropical and subtropical urban areas6, 7. Ae. albopictus has successfully expanded its geographical range and adopted itself to subtropical or even colder temperate zones3, 4, 8.
Very few arboviral vaccines are currently qualified or licensed for human use, including those for yellow fever, Japanese encephalitis and dengue (Dengvaxia® [CYD-TDV]). The two main limitations in arboviral vaccines are low coverage and their safety, for example an increased risk of complicated dengue (shock and hemorrhage) in seronegative individuals. Other arboviruses, including Zika and chikungunya, do not have any approved vaccines as per now. Novel approaches target mosquito salivary proteins (or their components) or use other non-pathogenic insect-specific viruses to induce immunity through cross-protection. Nanoparticles and mRNA – based vaccines are also in pre-clinical or non-human trials stage9, 10. The only option that remains in absence of a vaccine or drug, is vector control. Despite vector control interventions, an increase in the geographical spread of Aedes species has been observed, mainly due to urbanization, globalization, pesticide resistance and climate change11, 12, 13.
Understanding the mosquito microbiome is crucial in controlling and inhibiting viral transmission. The microbiome associated with mosquitoes plays a crucial role in their susceptibility to infection, their ability to become infectious, the replication of viruses in their gut, their immune response, and various ecological factors14, 15. Therefore, a whole microbiome analysis of mosquito microbiome can give insights about the vector’s capability in disease transmission and proposing effective and safe biological control strategies16, 17. Biological control strategies have already been used in vector-borne diseases including the use of Beauveria bassiana (entomopathogenic fungi), mostly found on the surface of water where mosquitoes breed kills both larvae and adults of many mosquito species18, 19. Introduction of some strains of alpha-proteobacteria Wolbachia into its non-natural host Ae. aegypti impacts its competence by causing cytoplasmic incompatibility and shortening its life span20, 21, 22. Identifying and disrupting the symbiotic relationships between mosquitoes and their natural symbionts, or genetically modifying mosquitoes to control pathogen’s replication and transmission can be used as a safe biological control approach23, 24, 25.
The current metagenomic study was conducted to explore the microbiome of Ae. aegypti and Ae. albopictus by taking advantage of advanced high throughput molecular techniques. Metagenomic sequencing of the Aedes microbiome will be performed to assess the diversity and relative abundance of its microbial community. Current research can be helpful for designing effective control of Aedes mosquitos and Aedes borne diseases based on their microbiome.
Materials and Methods
2
Sample collection and preparation
2.1
We obtained ethical approval of the study from the ethical committee of Department of Biosciences, COMSATS University Islamabad. This approval included the use of human blood for mosquito rearing, and human baiting to capture adult Aedes mosquitoes. We selected three dengue endemic regions of Pakistan for sample collection. We collected Ae. aegypti and Ae. albopictus from federal capital Islamabad, two districts of Punjab province (Rawalpindi and Lahore), and Swat district of Khyber Pakhtunkhwa from April-November 2022 (Fig. 1). We mainly collected immature life stages of these mosquitoes, however, when possible, we captured adults as well Fig. 2.Fig. 1. Sampling locations: Samples were collected from four regions, encompassing three geographical regions of Pakistan. The map is generated with the help of free and open-source software, qGIS (version 3.28). General Public License (GPL) developed by the Open-Source Geospatial Foundation Project (qgis.com).Fig. 2. Sampling immature and adult life stages of Aedes mosquitoes. A. Larvae collection from a water container. B. Ae. albopictus female trying to bite one of co-author during collection. C. Ae. aegypti male captured by our collaborator. D. Ae. aegypti female smashed in due course of collection.
Mosquitoes were maintained inside an incubator at the department of Biosciences, maintaining optimum temperature (28–30 °C) and relative humidity (70–80 %). Adult mosquitoes were provided with a 10 % sugar solution (ad libitum). We followed the WHO protocol to rear adults and larvae with minute modifications as our previous studies mention them26.
Morphological identification of adults was carried out by naked eye using morphological key defined by Rueda 200427. Female Aedes were separately pooled as 50 specimens/microtube and labelled properly. Samples were washed and cleaned with 70 % alcohol and treated with phosphate-buffered saline (PBS) and stored at −80 °C until nucleic acid extraction. Pools contained equal number of samples from all four locations.
DNA extraction
2.2
The mosquitos were mechanically homogenized with a pestle grinder within microtubes with nuclease free water and PBS for the preservation of nucleic acid. Samples were processed for DNA extraction using Qiagen® DNeasy Blood and Tissue kit (cat: 69504). The quantity of extracted DNA was measured using Qubit® 4 fluorometer (Invitrogen, Thermo Fisher, Scientific) to ensure its quality for the subsequent analysis. A negative control, without mosquito sample was run parallel to ensure the integrity of the whole extraction process.
Metagenomic sequencing and Bioinformatics analysis
2.3
DNA was then fragmented using suitable protocol and metagenomic library was prepared by attaching adapters to the DNA fragments to facilitate their binding and amplification. The library was then sequenced using high-throughput Illumina HiSeq instruments. After completion of sequencing, Illumina HiSeq/NovaSeq raw data was demultiplexed by index sequences, and paired end FASTQ files were generated for each sample. Adapter sequences and data with an average Phred quality score less than 20 were removed using Trimmomatic (v0.39) of the Knead data (v0.10.0) pipeline. Then, host-derived reads were filtered out by mapping them to the Ae. aegypti and Ae. albopictus reference genomes (GCF_002204515.2 and GCF_006496715.1, respectively) using Bowtie2 (v2.4.5). Flowchart details of library preparation is given in supplementary figure 1.
Assessment of sequencing depth and host depletion Efficacy
2.4
We filtered out host-derived reads by mapping the quality-controlled reads to the Ae. aegypti (RefSeq assembly accession: GCF_002204515.2) and Ae. albopictus (RefSeq assembly accession: GCF_006496715.1) reference genomes using Bowtie2 (v2.4.5) with default parameters. To quantify sequencing depth and the efficacy of this host depletion step, we report for each sample the raw read count, the number of reads passing adapter and quality trimming (Q-score > 30), and the number of reads that aligned to the respective host genome. The final non-host read count, which constitutes the input for all downstream microbiome analyses, is detailed in Table 1.Table 2..Table 1. Sequencing data and pre-processing metrics.Sample IDSpeciesRaw reads (n)QC Passed reads (n, %)Reads mapped to host (n, %)Non-host reads (n, %)Sample_01Ae. albopictus42,816,31840,235,501 (93.97 %)38,102,726 (94.7 %)2,132,775 (5.3 %)Sample_02Ae. aegypti39,093,17437,038,278 (94.74 %)35,086,441 (94.7 %)1,951,837 (5.3 %)Table 2. Bacterial genera identified in Ae. aegypti and Ae. albopictus.Percent (%) bacterial generaAe. aegyptiAe. AlbopictusKlebsiella52.630Pseudomonas88.152Acinetobacter34.740Brevundimonas11.266Enterobacter9.600Lampropedia0.582Stenotrophomonas3.021Pantoea0.002Elizabethkingia0.003Staphylococcus0.004
Taxonomic profiling of the microbiome
2.5
Taxonomic profiling was performed on the resulting non-host reads using MetaPhlAn4 (v4.0.0), which utilizes a library of clade-specific marker genes from ∼ 1 million microorganisms derived from NCBI reference genomes and species-level genome bins (SGBs). Briefly, the reads were mapped to this pan-microbial marker gene catalog using the integrated Bowti2 aligner. The relative abundance of each microbial species was calculated based on the average number of reads mapped to its unique marker genes. Any species for which less than 33 % of its marker genes were detected was considered absent and removed from the profile, as per the developer's recommendations.
Microbial diversity analysis
2.6
Microbial diversity (including richness, Shannon and Simpson values) was calculated using species abundance from the ‘calculate_diversity.R’ script of MetaPhlAn4. In addition, weighted and unweighted UniFrac distances, which are beta diversity values, were calculated based on the phylogenetic tree pre-calculated by the MetaPhlAn4 database. This whole process generated an extensive report of taxonomic classification of both bacteria and DNA viruses in Ae. aegypti and Ae. albopictus pools.
Results
3
DNA from two pools of Aedes mosquitoes (Ae. aegypti and Ae. albopictus) was successfully extracted. Concentrations from both samples were confirmed via Qubit® Fluorometer quantification, meeting the necessary criteria for metagenomic sequencing28. From these samples, a total of 15.6 gigabases of raw sequence data was generated. Following quality filtering and the removal of host-derived reads, a total of 12.1 Gb of reads were retained, which were then used for detailed microbiome analysis.
Relative microbial abundance in Ae. Aegypti and Ae. Albopictus
3.1
Metagenomic analysis revealed the pattern of bacteria population in both pools (Fig. 3), table 2. Proteobacteria emerged as the most abundant phylum which displayed more than ∼ 99.99 % in Ae. aegypti to 100 % in Ae. albopictus. In comparison, Firmicutes and Bacteroidota were only found in Ae. aegypti with overall abundance less than 1 %.Fig. 3. The relative abundance of bacterial genera in Ae. aegypti and Ae. albopictus. The most abundant are Acinetobacter and Pseudomonas in Ae. aegypti and Ae. albopictus respectively. Klebsiella and Enterobacter are in moderate abundance observed only in Ae. aegypti while other genera including Elizabethkingia, Pantoea, Staphylococcus and Stenotrophomonas are in low abundance.
Each species of mosquito sheltered unique genera of bacteria as; Klebsiella, Stenotrophomonas, Acinetobacter, Staphylococcus, Elizabethkingia, Pantoea and Enterobacter unique to Ae. aegypti. Similarly, Pseudomonas, Brevundimonas, and Lampropedia were unique to Ae. albopictus. Importantly, recently characterized bacterial specie Lampropedia aestuarii was identified for the first time in Ae. albopictus in the current study. Host-specific adherence of microbial population and diversity are likely to be due to the combination of host-specific factors, ecological interactions and environmental conditions16.
The detailed analysis regarding the identified species within each mosquito revealed that in the Ae. aegypti sample, the genera Acinetobacter and Klebsiella were the most abundant. Specifically, the most common species were Acinetobacter baylyi (30.68 %), Klebsiella pneumoniae (30.79 %), and Klebsiella variicola (19.74 %), followed by Acinetobacter soli (3.98 %), Klebsiella quasipneumoniae (2.1 %), and Acinetobacter bereziniae (0.08 %) (Fig. 4). In Ae. albopictus three species of genera Pseudomonas, Brevundimonas and Lampropedia were observed and P. putida emerged as the most abundant species (88.15 %) while L. aestuarii (1 %) was observed as the least abundant species as shown in Fig. 4.Fig. 4. The relative abundance of bacterial species in Ae. aegypti and Ae. albopictus. The most abundant are Acinetobacter baylyi and Pseudomonas putida in Ae. aegypti and Ae. albopictus respectively. Klebsiella species and Enterobacter hormaechei are observed in moderate abundance only in Ae. aegypti Brevundimonas diminuta and Lampropedia aestuarii have moderate to low abundance in Ae. albopictus only.
Microbial Alpha diversity
3.2
Microbial alpha diversity measures species diversity in a specific ecosystem or sample. In the context of Aedes, microbial Alpha Diversity refers to the microbiomes associated with Ae. aegypti and Ae. albopictus. Microbial Alpha Diversity was measured by the Shannon Diversity Index, Observed Species Index, and Simpson Index (λ). Table 3 shows microbial diversity in the sample pool of Ae. aegypti and Ae. albopictus. The Observed Species Index refers to the number of distinct species present within each sample. It disregards the relative abundance of each species. A sample with a high value for the Observed Species Index demonstrates increased stability, imparting enhanced resistance against disturbances29, 30. The Simpson Index (λ) measures dominance, where a value of 1 indicates complete dominance by a single species (low diversity) and values approaching 0 indicate higher diversity.Table 4..Table 3. Bacterial Alpha Diversity.Sample poolsObserved species IndexShannon Diversity index(H)Gini Simpson IndexAedes aegypti121.40.73Aedes albopictus30.390.21Table 4Relative abundance (%) of prevalent virus species across Ae. aegypti and Ae. albopictus sample pools.GenusSpeciesAe. aegyptiAe. albopictusSiphoviridae**Escherichia phage HK63970.960Hk97virus**Enterobacterial phage mEp39022.560Myoviridae**Cronobacter phageENT476703.020Epsilon15virus**Salmonella virus Epsilon151.20Siphoviridae**Enterobacteria phage mEp2371.160N15virus**Escherichia virus N151.10Siphoviridae**Lactobacillus phage A2062.95Retroviridae**Human endogenous retrovirus K037.05
In our results, Ae. aegypti exhibited greater microbial diversity than Ae. albopictus, as evidenced by a higher Observed Species Index and Shannon Diversity Index, and a lower Simpson Index (λ).
In contrast, lower value of Observed Specie Index in Ae. albopictus, comprising three (03) species, suggests a microbial population that is relatively less diverse. Similarly, Shannon Diversity Index (H) considers both species diversity and evenness within an ecosystem or sample31. Table 3 shows that Ae. aegypti sample pool has bigger value (1.4), compared to Ae. albopictus sample pool further highlight the diversity and evenness in Ae. aegypti. Simpson’s index refers to the measurement of probability that two individuals selected randomly from a sample are related to the same species32. The lower value of Simpson Index shows higher microbial diversity. Higher value of Simpson Index (0.73) within Ae. aegypti signify lesser microbial diversity relative to Ae. albopictus33.
Relative abundance of DNA viruses?
3.3
The presence of viruses in Aedes pose greater concern both for ecosystem and public health. The relative abundance (Fig. 5, table 4) of virus communities shows the presence and distribution of different DNA viruses within both sample pools. Genus Siphoviridae is predominant genus with 69.96 % abundance in Ae. aegypti and 62.95 % abundance in Ae. albopictus, Hk97, Myoviridae, Epsilon15, Siphoviridae and N15 are the genera unique to Ae. aegypti while Retroviridae is the genus unique to Ae. albopictus. Furthermore, relatively significant abundant species Human endogenous retrovirus K belonging to the genus Retroviridae has been observed for the first time in Ae. albopictus. This is another novel finding of the current study which can make ground to explore vector competence of Ae. albopictus.Fig. 5. Relative abundance and percentage of DNA viral phages in Ae. aegypti and Ae. albopictus, with the most abundant phage Escherichia_phage_HK639(70.96%) and Lactobacillus_Phage_A2(62.95%) in both Ae. aegypti and Ae. albopictus respectively.
Discussion
4
Understanding microbiota of an organism which transmits parasites and their mutual interactions makes is extremely important. Therefore, we aimed to identify and compare the microbiota of two arbovirus vectors, Ae. aegypti and Ae. albopictus in Pakistan and its role in vector’s biology and competence for pathogen replication and transmission.
Metagenomic sequencing is a rapid and highly sensitive technique which can detect whole microbiomes in a pool of mixed community without any prior knowledge so, the genetic materials of all organisms, including bacteria, viruses, fungi, and archaea are isolated and screened in a single step28, 34. It is crucial to identify associated microbiota of mosquito species and its possible role in designing effective and safe biological vector control strategies.
We identified that Proteobacteria was the most abundant phylum found in both samples of Ae. aegypti and Ae. albopictus which align with previous studies35. Phylum Firmicutes and Bacteroidetes existed in low abundance and were unique only in Ae. aegypti35, 36, 37. But in our study Firmicutes and Bacteroidetes were not found in Ae. albopictus. It can be assumed that protobacteria is conserved microbiota of both Aedes species.
In Proteobacteria, two genera, Pseudomonas and Klebsiella, were abundant in our samples. Pseudomonas stimulates the immune system and produces antimicrobial peptides which might help them fight against viruses38. Klebsiella produce hemolysin, facilitate the digestion of food after absorption of blood39, 40. The most abundant bacterial species was Klebsiella pneumoniae and Pseudomonas putida in Ae. aegypti and Ae. albopictus respectively*. Klebsiella* species has been identified as an efficient symbionts and screened in all stages of life cycle of Aedes mosquitoes, playing an important role in larval growth and development41. Mosquera et al has also indicated that these bacterial species are transmitted by female mosquito while oviposition and larvae eventually acquire them after hatching. This survival and supportive behavior of vector might indicate significant role of Klebsiella species. Paul et al identified an enzyme, chitinase in one of the newly identified strain of Pseudomonas putida with effective role as biopesticide. Chitin is part of integument in larval as well as in adults of mosquitos42, 43. In this study, the presence of Pseudomonas putida as part of the Aedes microbiome should be further investigated to determine its role in larvicidal activities. A total of fifteen bacterial species were isolated from both samples; most were gram-negative, with the exception of one, Staphylococcus hominis. In contrast to previous studies, this gram-positive bacteria was found in lab raised Ae. aegypti44. Lampropedia aestuarii was observed for the first time in Ae albopictus in our study.
Among viruses, Escherichia phage HK639 was dominantly found Ae. aegypti and Lactobacillus phage in Ae. albopictus. Another notable finding of this study was the presence of Human endogenous retrovirus K in Ae. albopictus. The current study identified the Siphoviridae as the predominant family in the DNA virome in both mosquitoes which aligns with the previous studies, demonstrating that phages belonging to Siphoviridae are abundant in mosquitoes45, 46. However, the current study observed that both pools have variable viral communities, unique to each of the Ae. aegypti and Ae. albopictus mosquito. We identified Human endogenous retrovirus type-K (HERV-K) for the first time in Ae. albopictus mosquito. Currently this group of viruses is of great interest for researchers due its association with other human diseases. HERV detected in mosquito samples might be of human origin as the wild type mosquitoes feed on human blood, however, further investigations are needed to confirm if HERV exists naturally as part of the mosquitoes’ genome. Moreover, HERV was detected only in Ae. albopictus but not in Ae. aegypti. HERVs comprising 8 % of the human genome, are the remnants of the ancient germline with integrated retroviruses47, 48. Although HERVs are poorly explored, there are evidence defining their pathological roles in various diseases including autoimmune, cancer, and neurodegenerative diseases. HERV-K expression had been shown to be associated with the progression and mortality of SARS-CoV-247, 49, 50. So, the presence of HERVs in Ae. albopictus might have a contributing role in mosquito competency and can be marker in disease severity.
Fifteen different species of bacteria were isolated from both Aedes species*.* Species related variations in microbial populations are attributed to the period and diet of the host mosquito51, 52. The microbiome of different mosquitoes species including Ae. albopictus and Ae. aegypti also affect their competence, and antagonism to some arboviruses53, 54, 55. The presence of diverse microbial compositions within Ae. aegypti mosquito population has significant implications on the rate of infection in different ecological and geographical contexts. Here in this study the Shannon index indicating unique microbial diversity of both mosquito species although both were collected from same geographical locations.
In this study, certain strains found in each sample exhibit distinct properties, showing that localized interactions might have a role in the susceptibility of mosquitoes to different pathogens. However, species that generally exist serve as primary indicators of the crucial microbiome characteristics that have a universal impact on the interaction between mosquitoes and pathogens. The prevalence rate of these species influences the microbial dynamics at a locality, impacting infection vulnerability. The observed fluctuations in bacterial and viral composition and prevalence indicate that each sample pool corresponds to different ecological behavior as both were sharing the common habitat. The observed variation in the rate of infectivity may be attributed to variation in ecological niche thereby provides an additional insight into the observed gap. Nonetheless, this information alone is not enough to find out their roles, interactions, or significant implications in the ecosystem. Further studies required to find out the impact of microbiota of these two particular vector species and its role in efficient replication and transmission of pathogens. But a comprehensive understanding of the subject matter can be used to improve the potency of a particular disease control program.
These observations show that ecological interactions or environmental factors might favor the growth and proliferation of specific microbial populations. The existence of the diverse microbial population within Ae. aegypti will have significant implications for apprehending the microbiome56, 57. Variation in microbial population may have an impact on the varying rates of infection in different geographical areas.
The microbiota of Aedes mosquitoes impact its fitness, competence and biological competition58. The microbiome of habitat and genetic diversities of mosquitoes impact how well and adult mosquito can be infected of virus and can potentially transmit it59. Invading pathogen has to interact with gut microbiome of adult mosquito. Therefore, impacting its replication. Few species of bacterial genera Pseudomonas has shown in in vitro studies that they have potential to inhibit viral replication by making antiviral metabolites in Ae. albopictus60. In our study we have identified genus Pseudomonas in Ae. albopictus which need further research to find its role as an antiviral agent.
Few studies have shown that microbiota of Ae. aegypti has a significant role for its susceptibility and transmission to DENV-261. Similarly, Ae. albopictus is rapidly spreading in all continents and is a competent vector for dengue virus. Most of the studies are focusing on interaction of mosquitoes naturally occurring symbiont Wolbachia and its immunity but the impact of whole microbiome on its immune response and driving vector’s competence are less explore62, 63. Many studies are focusing on exploring the potential role of microbiome in insecticide resistance as this phenomenon is not only vertically transmitted but also maintained at gene level (Sahar Fazal et al, 2023, role of mosquito microbiome in Insecticide Resistance).
Conclusion
5
Ae. aegypti and Ae. albopictus are spreading to new geographical areas and thus becoming a substantial risk for human health. Their microbiota plays a critical role in shaping their immune system, competence and severity of disease transmission. Variation in microbiota is corelated with variation in competence of both species. The current study can help to suggest new microbial agents for the control and elimination of dengue vectors. These findings will help to design more effective and environmentally friendly vector control agents.
Data Availability
6
The raw sequencing data generated during this study are not publicly available due to restrictions in the original data sharing agreement associated with the sequencing service.. However, the processed data essential for reproducing all analyses and conclusions are provided within the main text or Supplementary Information of this article. This includes the relative abundance profiles of all detected microbial species and the detailed sequencing metrics and read counts for each sample.
CRediT authorship contribution statement
Shahzadi Asia Nadeem: Writing – review & editing, Resources, Methodology, Investigation, Formal analysis. Ijaz Ali: Supervision, Methodology, Conceptualization. Hazrat Hussain: Writing – original draft, Methodology. Ihsan Ullah: Supervision, Methodology. Wajid Ali: Validation, Conceptualization. Khalid J. Alzahrani: Funding acquisition. Hamid Ali: Resources. Zarak Imtiaz Khan: Software, Methodology. Kasim Sakran Abass: Funding acquisition. Rafi ur Rahman: Writing – original draft, Supervision, Software, Methodology, Formal analysis, Conceptualization.
Declaration of Competing Interest
The authors declare the following financial interests/personal relationships which may be considered as potential competing interests: Ijaz Ali reports administrative support and writing assistance were provided by COMSATS University Islamabad. If there are other authors, they declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Guzman M.G.Harris E.Dengue The Lancet 3859966201545346510.1016/S 0140-6736(14)60572-925230594 · doi ↗ · pubmed ↗
- 2Jones K.E.Patel N.G.Levy M.A.Global trends in emerging infectious diseases Nature 4517181200899099310.1038/nature 0653618288193 PMC 5960580 · doi ↗ · pubmed ↗
- 3Kraemer M.U.G.Sinka M.E.Duda K.A.The Global Distribution of the Arbovirus Vectors Aedes Aegypti and Ae.albopictus. 1–18201510.7554/e Life.08347 PMC 449361626126267 · doi ↗ · pubmed ↗
- 4Kraemer M.U.G.Sinka M.E.Duda K.A.The Global Distribution of the Arbovirus Vectors Aedes Aegypti and Ae.albopictus. 1–18201510.7554/e Life.08347 PMC 449361626126267 · doi ↗ · pubmed ↗
- 5Leta S.Beyene T.J.De Clercq E.M.Amenu K.Kraemer M.U.G.Revie C.W.Global risk mapping for major diseases transmitted by Aedes aegypti and Aedes albopictus International Journal of Infectious Diseases : IJID : Official Publication of the International Society for Infectious Diseases 672018253510.1016/j.ijid.2017.11.02629196275 PMC 5976855 · doi ↗ · pubmed ↗
- 6Espinal M.A.Andrus J.K.Jauregui B.Emerging and Reemerging Aedes-Transmitted Arbovirus Infections in the Region of the Americas: Implications for Health Policy American Journal of Public Health 1093201938739210.2105/AJPH.2018.30484930676796 PMC 6366516 · doi ↗ · pubmed ↗
- 7Souza-Neto J.A.Powell J.R.Bonizzoni M.Aedes aegypti vector competence studies: a review Infection, Genetics and Evolution : Journal of Molecular Epidemiology and Evolutionary Genetics in Infectious Diseases 67201919120910.1016/j.meegid.2018.11.00930465912 PMC 8135908 · doi ↗ · pubmed ↗
- 8Paupy C.Delatte H.Bagny L.Corbel V.Fontenille D.Aedes albopictus, an arbovirus vector: from the darkness to the light Microbes and Infection 1114–1520091177118510.1016/j.micinf.2009.05.00519450706 · doi ↗ · pubmed ↗