ALK‐Positive Histiocytosis With Unilateral Breast Involvement: A Case Report
Xuechun Liu, Dong Ren, Yanfang Liang

TL;DR
A rare case of ALK-positive histiocytosis in the breast was diagnosed and successfully treated with surgery alone.
Contribution
This case report highlights the rare occurrence of ALK-positive histiocytosis in the breast and its successful management with surgical resection.
Findings
ALK-positive histiocytosis can present as a painless breast mass and is diagnosed using histopathology and immunohistochemistry.
Complete surgical resection without adjuvant therapy resulted in no recurrence after 15 months of follow-up.
Accurate diagnosis requires a combination of histopathology, immunohistochemistry, and molecular testing to differentiate from other histiocytic disorders.
Abstract
APH is a rare disorder characterized by the proliferation of ALK‐expressing histiocytes with variable anatomical involvement; however, mammary involvement is exceptionally rare. A 32‐year‐old woman presented with a painless right breast mass. Ultrasound identified a 9 × 8 mm hypoechoic nodule, categorized as BI‐RADS 4A. Microscopy showed spindle cells in whorled patterns with characteristic nuclear folding. Immunohistochemistry demonstrated positivity for CD68, CD163, and ALK, and negativity for S‐100, CD1a, and CD207. The Ki‐67 proliferation index was approximately 10%. Fluorescence in situ hybridization (FISH) confirmed ALK gene rearrangement (The partner gene not identified), establishing the diagnosis of APH. The patient underwent complete resection without adjuvant therapy, and no recurrence was observed at the 15‐month ultrasound follow‐up. Accurate diagnosis of APH requires…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
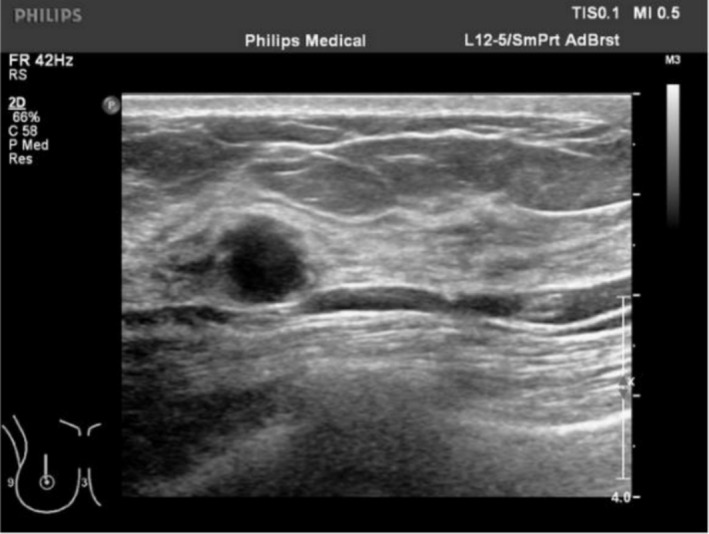 FIGURE 1
FIGURE 1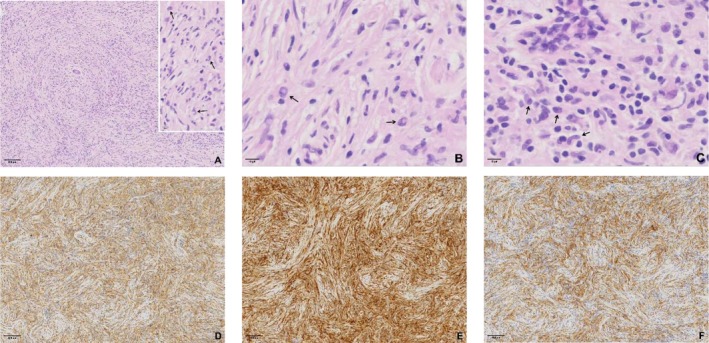 FIGURE 2
FIGURE 2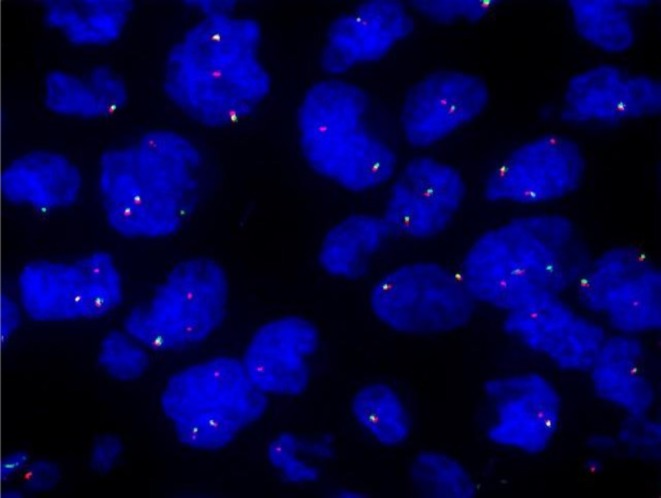 FIGURE 3
FIGURE 3| First author, year | Age (years) | Ethnicity | Tumor size (mm) | ALK fusion partner | Treatment | Follow‐up (months) |
|---|---|---|---|---|---|---|
| Osako, 2022 [ | 38 | Japanese | 17 | KIF5B (24)‐ALK (20) | Resection | No evidence of disease (11 months) |
| Osako, 2022 [ | 45 | Japanese | 13 | KIF5B (24)‐ALK (20) | Resection | No evidence of disease (13 months) |
| Zhou, 2023 [ | 46 | Hispanic | 20 | KIF5B‐ALK | Crizotinib | No evidence of disease (4 months) |
| Kurita, 2022 [ | 38 | NA | 12 | KIF5B‐ALK | NA | NA |
| Chang, 2019 [ | 40 | Chinese | NA | KIF5B‐ALK | Resection | No evidence of disease (42 months) |
| Kashima J, 2021 [ | 45 | Asian | 16 | KIF5BALK | Resection | No evidence of disease (1 months) |
| Kashima, 2021 [ | 16 | Asian | 21 | KIF5BALK | Resection, alectinib | No evidence of disease (44 months) |
| The present case | 32 | Chinese | 9 | ALK gene rearrangement(FISH) | Resection | No evidence of disease (15 months) |
| Case | Sex | Age | Affected site | Histopathological features |
|---|---|---|---|---|
| 1 | Male | 2 days postnatal | Liver | Sinusoidal infiltration pattern with compression of adjacent hepatocytes and variable portal tract involvement [ |
| 2 | Female | 7 and 10 years old | Cerebellar vermis, central cortex | Sheets of large epithelioid cells with irregularly folded nuclei, fine chromatin, foamy cells, Touton giant cells, and focal emperipolesis [ |
| 3 | Male | 38 years | Mediastinum | Large histiocytes with abundant eosinophilic cytoplasm, cleaved or lobulated nuclei, and small nucleoli (meningothelial‐like morphology). Scattered foamy cells, small lymphocytes, rare Touton giant cells, and emperipolesis [ |
| 4 | Male | 49 years | Lymph node | Polygonal histiocytes (medium to large) with moderate eosinophilic cytoplasm, fine chromatin, inconspicuous nucleoli, and frequent nuclear grooves or folds. Focal pleomorphic cells with prominent nucleoli and scattered Touton giant cells [ |
| 5 | Female | 52 years | Lung | Round to spindle‐shaped cells with abundant eosinophilic vesicular cytoplasm, mild nuclear atypia, and indistinct nucleoli. Background of foamy histiocytes, lymphocytes, and hemosiderin deposits [ |
- —Guangdong Basic and Applied Basic Research Foundation10.13039/501100021171
- —High Level Scientific Research Incubation Foundation of Binhaiwan Central Hospital of Dongguan
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsHistiocytic Disorders and Treatments · Tuberous Sclerosis Complex Research · Amyloidosis: Diagnosis, Treatment, Outcomes
Introduction
1
APH is a rare disorder characterized by the proliferation of morphologically distinctive histiocytes expressing ALK protein. Since its initial description by Chan et al. in 2008 [1], numerous cases have been reported worldwide. The 2022 WHO Classification of Haematolymphoid Tumors now formally recognizes APH as a distinct entity [2]. Although APH can involve various anatomical sites—including the lung, skin, and central nervous system [3, 4, 5, 6, 7], mammary involvement remains exceptionally rare(Table 1). This report presents a rare case of mammary APH and summarizes its clinicopathological features to aid in the recognition and understanding of this uncommon entity.
Case Presentation
2
A 32‐year‐old woman presented in December 2023 with a painless right breast mass. Physical examination revealed no erythema, swelling, ulceration, or skin changes in the breast and no abnormalities at other sites. Laboratory tests including complete blood count and biochemistry were within normal limits. The patient underwent breast ultrasound and pathological examination, with no systemic staging imaging performed. Breast ultrasound identified a 9 × 8 mm hypoechoic nodule at the 12 o'clock position, located 33 mm from the nipple. The lesion exhibited heterogeneous echogenicity, circumscribed margins, an aspect ratio greater than 1, and no detectable vascularity (CDFI Grade 0). It was classified as BI‐RADS 4A (Figure 1). Gross examination of the excised tissue revealed fragmented grayish‐white to yellow material measuring 20 × 20 × 20 mm (Adjacent normal tissue included via a Mammotome biopsy). Histologic evaluation showed spindle cells arranged in whorled patterns, with abundant pale cytoplasm and vesicular nuclei featuring characteristic doughnut‐shaped or reniform folding (Figure 2). Additional findings included Touton giant cells, focal inflammatory infiltrates, and stromal hyperplasia. Immunohistochemistry demonstrated positivity for CD68, CD163, and ALK, and negativity for S‐100, CD1a, and CD207. The Ki‐67 proliferation index was approximately 10% (Figure 2). FISH confirmed ALK gene rearrangement (Figure 3), establishing the diagnosis of APH.
Ultrasound findings: A hypoechoic nodule in the right breast (BI‐RADS category 4A).
Histologic features of ALK‐positive histiocytosis. (A) Spindle‐shaped cells arranged in whorled patterns (HE, ×40). Horseshoe‐shaped and bean‐like or reniform folding nuclei (→, HE, ×400). (B) Doughnut‐shaped histiocytic nuclei (→, HE, ×400). (C) Characteristic nuclear folding and nuclear grooves (→, HE, ×400). (D) CD68 cytoplasmic positivity (EnVision method, ×40). (E) CD163 cytoplasmic positivity (EnVision method, ×40). (F) ALK cytoplasmic positivity (EnVision method, ×40).
ALK gene rearrangement confirmed by FISH.
All tissue samples were fixed in 10% neutral buffered formalin, embedded in paraffin, and sectioned into 4 μm slices for histological examination by hematoxylin–eosin (HE) staining and immunohistochemical evaluation of tumor cell immunophenotypes. ALK monoclonal antibody and Ki‐67 detection were performed by Gene Biotechnology, while CD68, CD163, Calponin, Desmin, SMA, P‐CK, S‐100, CD34, CD1a, and CD207 were analyzed by Maixin Biotechnology. ALK gene rearrangements were detected by FISH using probes from Abbott Gene Tech (Shanghai).
Differential Diagnosis, Investigations and Treatment
3
Histiocytes in APH typically express histiocytic markers such as CD68, CD163, lysozyme, and ALK. They lack expression of CD1a and CD207. However, a reported case of APH was positive for S100 and dendritic cell markers (fascin, Factor XIIIa), yet still negative for both CD1a and Langerin [1]. The differential diagnosis of APH includes other histiocytic disorders, most notably Erdheim‐Chester disease (ECD), Langerhans cell histiocytosis (LCH), and Rosai‐Dorfman disease (RDD).
ECD typically affects patients aged 55–60 and involves the bones, cardiovascular system, and retroperitoneum. It is characterized by foamy mononuclear histiocytes with small nuclei, frequent multinucleated or Touton‐type giant cells, and variable fibrosis. Over 50% of ECD cases harbor BRAF V600E mutations [11]. In contrast to APH, ECD features foamy histiocytes and lacks ALK rearrangements, although rare ALK expression without rearrangement has been reported [12].
LCH usually presents with localized bone pain or tumor‐like lesions and is characterized by cells resembling Langerhans cells that are actually myeloid precursors sharing their immunophenotype [13]. These cells are positive for CD1a, CD207, and S100, and contain pathognomonic Birbeck granules on ultrastructural analysis. Approximately 50% of LCH cases also carry BRAF V600E mutations, differing from APH, which consistently shows ALK rearrangements and is negative for CD1a and CD207 [14].
RDD is characterized by sinusoidal expansion by large histiocytes with abundant cytoplasm, prominent nucleoli, and the presence of emperipolesis. RDD cells are positive for S100, CD163, and CD68, but negative for CD1a [15]. Unlike the sinusoidal growth pattern of RDD, mammary APH typically demonstrates a spindle cell morphology, aiding in its distinction.
Spindle cell morphology in APH may also mimic inflammatory myofibroblastic tumor (IMT), which expresses SMA and desmin but lacks histiocytic markers. Although around 50% of IMTs exhibit ALK expression, they often harbor TPM3‐ALK, TPM4‐ALK, or CLTC‐ALK fusions, distinct from the KIF5B‐ALK fusion predominantly seen in APH [11].
The patient in this case underwent complete surgical resection without adjuvant therapy. ALK inhibitors represent the primary treatment modality for APH, often achieving durable clinical remission, although consensus on their use as first‐line versus for refractory cases remains lacking. Notably, a reported case of ALK‐positive ECD‐like histiocytosis arising in the setting of chronic lymphocytic leukemia/small lymphocytic lymphoma (CLL/SLL) demonstrated a favorable response to the Bruton tyrosine kinase (BTK) inhibitor ibrutinib, suggesting potential therapeutic utility of BTK inhibition in select cases [16]. Molecular profiling remains essential for guiding targeted therapy and optimizing individualized treatment strategies.
Conclusions and Results (Outcome and Follow‐Up)
4
The patient showed no evidence of recurrence at the 15‐month ultrasound follow‐up. APH is generally associated with a favorable prognosis, with most patients achieving stable disease or complete remission following systemic chemotherapy, surgical resection, or targeted therapy. However, certain cases may exhibit aggressive histological features—for example, a reported mesenteric case demonstrated intravascular invasion involving large veins [17]. Due to the rarity of APH, the correlation between histologic features and clinical behavior remains poorly defined, underscoring the need for further investigation. Additionally, treatment‐related complications may significantly impact outcomes and warrant careful management. In the present case, serial ultrasound examinations confirmed disease stability without evidence of progression or metastasis, in line with the overall indolent biological behavior of this tumor.
Discussion
5
APH presents notable diagnostic challenges, with multiple cases reported across Asian populations, particularly in China and Japan, spanning a broad age range from 2 months to 66 years. This entity most commonly involves the central nervous system, skin, and lungs, while mammary involvement remains exceptionally rare. Current evidence suggests that mammary APH primarily affects Asian females aged 30–40 years, though no clear clinicopathological definition has been established to date [8]. Cutaneous presentations typically appear as papules or nodules, in contrast to mammary lesions, which are often asymptomatic and incidentally detected on imaging, as demonstrated in the present case.
Histopathological features of APH demonstrate considerable morphological diversity. It is typically composed of large histiocytes with abundant eosinophilic cytoplasm and irregular nuclei characterized by delicate chromatin and complex nuclear folding patterns, distinguishing it from other histiocytic proliferations. In some cases, the lesion comprises uniform spindle cells arranged in fascicular or storiform patterns, often accompanied by lymphocytic infiltration, which can mimic inflammatory disorders or proliferative lesions such as inflammatory myofibroblastic tumor (IMT). Notably, spindle cell morphology is frequently observed in mammary APH, while it remains relatively uncommon in APH at other anatomical sites [8]. Our case is consistent with previously reported features of mammary APH, including prominent spindle cell proliferation, with subtle histological variation across anatomical sites as summarized in recent literature (Table 2).
Recent molecular advances have begun to elucidate the genomic landscape of histiocytic disorders. ALK gene rearrangement is considered the defining molecular hallmark of APH and a potential oncogenic driver. While ALK fusions occur with various partners across tumors, specific fusion partners tend to be enriched in particular disease entities. In APH, KIF5B‐ALK is the most frequently reported fusion, though rare variants include TPM3‐ALK, COL1A2‐ALK, TRIM33‐ALK, CLTC‐ALK, TFG‐ALK, DCTN1‐ALK, and EML4‐ALK [3].
KIF5B‐ALK–positive cases typically display sheet‐like histiocytic growth composed of cells with abundant eosinophilic cytoplasm, irregular nuclear contours (including folding, lobulation, and clefting), finely dispersed chromatin, and inconspicuous nucleoli.
TRIM33(exon12)‐ALK(exon20) fusions are associated with nested histiocytic proliferation within a delicate capillary network, occasionally exhibiting hemangiopericytoma‐like vascular patterns. Histiocytes show indistinct cytoplasmic borders, moderately eosinophilic cytoplasm, and ovoid nuclei arranged in alveolar patterns, with prominent basophilic nucleoli and no nuclear grooves. Mammary APH frequently demonstrates lymphoid follicles with plasma cells, eosinophils, and lymphocytes, contributing to IMT‐like histologic features [17].
COL1A2(exon51)‐ALK(exon19) fusions are typically associated with cutaneous or soft tissue lesions, characterized by distinct histiocytic islands rather than sheets. The neoplastic cells exhibit amphophilic cytoplasm and monotonous nuclei with single prominent nucleoli [17].
Author Contributions
Xuechun Liu: conceptualization, data curation, investigation, project administration, visualization, writing – original draft, writing – review and editing. Dong Ren: writing – review and editing. Yanfang Liang: funding acquisition, supervision, writing – review and editing.
Funding
This work was supported by the Guangdong Basic and Applied Basic Research Foundation, 2021B1515140066. High Level Scientific Research Incubation Foundation of Binhaiwan Central Hospital of Dongguan, 2024002.
Consent
The patient provided written informed consent for the publication of this case report, in accordance with the journal's consent policy.
Conflicts of Interest
The authors declare no conflicts of interest.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1J. K. Chan , L. Lamant , E. Algar , et al., “ALK+ Histiocytosis: A Novel Type of Systemic Histiocytic Proliferative Disorder of Early Infancy,” Blood 112, no. 7 (2008): 2965–2968, 10.1182/blood-2008-03-147017.18660380 · doi ↗ · pubmed ↗
- 2J. D. Khoury , E. Solary , O. Abla , et al., “The 5th Edition of the World Health Organization Classification of Haematolymphoid Tumours: Myeloid and Histiocytic/Dendritic Neoplasms,” Leukemia 36, no. 7 (2022): 1703–1719, 10.1038/s 41375-022-01613-1.35732831 PMC 9252913 · doi ↗ · pubmed ↗
- 3W. Liu , H. J. Liu , W. Y. Wang , et al., “Multisystem ALK‐Positive Histiocytosis: A Multi‐Case Study and Literature Review,” Orphanet Journal of Rare Diseases 18, no. 1 (2023): 53, 10.1186/s 13023-023-02649-x.36915094 PMC 10010018 · doi ↗ · pubmed ↗
- 4H. Huang , G. Gheorghe , P. E. North , and M. Suchi , “Expanding the Phenotype of ALK‐Positive Histiocytosis: A Report of 2 Cases,” Pediatric and Developmental Pathology 21, no. 5 (2018): 449–455, 10.1177/1093526617740784.29224419 · doi ↗ · pubmed ↗
- 5L. Qiu , S. P. Weitzman , L. J. Nastoupil , M. D. Williams , L. J. Medeiros , and F. Vega , “Disseminated ALK‐Positive Histiocytosis With KIF 5B‐ALK Fusion in an Adult,” Leukemia & Lymphoma 62, no. 5 (2021): 1234–1238, 10.1080/10428194.2020.1861273.33353436 · doi ↗ · pubmed ↗
- 6Y. Bai , W. Sun , D. Niu , et al., “Localized ALK‐Positive Histiocytosis in a Chinese Woman: Report of a Case in the Lung With a Novel EML 4‐ALK Rearrangement,” Virchows Archiv 479, no. 6 (2021): 1079–1083, 10.1007/s 00428-021-03092-8.33825946 · doi ↗ · pubmed ↗
- 7C. G. Lucas , A. Gilani , D. A. Solomon , et al., “ALK‐Positive Histiocytosis With KIF 5B‐ALK Fusion in the Central Nervous System,” Acta Neuropathologica 138, no. 2 (2019): 335–337, 10.1007/s 00401-019-02027-7.31119374 PMC 6712982 · doi ↗ · pubmed ↗
- 8T. Osako , A. Kurisaki‐Arakawa , A. Dobashi , et al., “Distinct Clinicopathologic Features and Possible P‐Athogenesis of Localized ALK‐Positive Histiocytosis of the Breast,” American Journal of Surgical Pathology 46, no. 3 (2022): 344–352, 10.1097/PAS.0000000000001794.34482333 · doi ↗ · pubmed ↗