Research Progress of Ferroptosis in Cerebral Infarction
Yilan Fei, Qi Leng

TL;DR
This paper reviews how a type of cell death called ferroptosis contributes to brain injury after stroke and explores new treatment strategies to prevent it.
Contribution
The paper provides a comprehensive review of ferroptosis mechanisms and emerging therapies in cerebral infarction, highlighting translational opportunities.
Findings
Ferroptosis in cerebral infarction is driven by glutathione depletion, GPX4 inactivation, and iron-catalyzed lipid peroxidation.
Therapeutic strategies like ferroptosis inhibitors, iron chelators, and RNA/gene-targeted interventions show strong neuroprotection in preclinical models.
Biomarkers such as oxylipins and GPX4 fragments may help identify patients who could benefit from ferroptosis-targeted therapies.
Abstract
Purpose: To synthesize current mechanistic insights and translational progress on ferroptosis, a regulated, iron‐dependent, nonapoptotic cell death pathway in the pathophysiology and treatment of cerebral infarction (ischemic stroke), and to outline therapeutic opportunities and remaining gaps for clinical application. Method: Narrative, focused review of preclinical and translational studies (in vitro, ex vivo, and in vivo ischemia/reperfusion and middle cerebral artery occlusion models), alongside emerging biomarker, nanocarrier, and gene/RNA‐based strategies reported up to 2025. Evidence was organized across five domains: (1) redox and lipid peroxidation biology; (2) iron metabolism and ferritinophagy; (3) mitochondrial dysfunction; (4) neuroinflammation and blood–brain barrier integrity; and (5) therapeutic development and early clinical exploration. Finding: Ferroptosis in…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
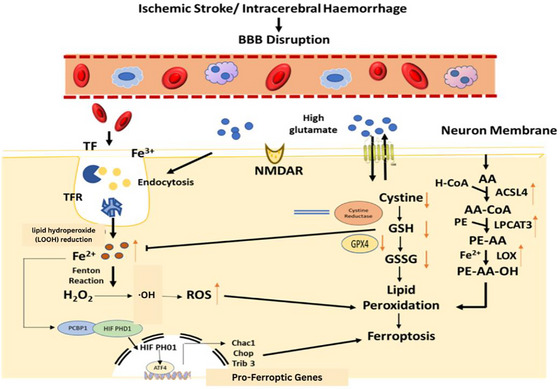 FIGURE 1
FIGURE 1| S. No. | Research area | Key insights and mechanisms | Significance | References |
|---|---|---|---|---|
| 1 |
| Mitochondria act as key amplifiers of ferroptosis under ischemic conditions. Ischemia−reperfusion leads to mitochondrial dysfunction, excessive ROS generation, and disruption of iron–sulfur clusters, which accelerates lipid peroxidation in neuronal cells. | Reveals a mitochondria‐centric pathway in ferroptosis initiation and suggests targeting mitochondrial redox balance. | Fu et al. ( |
| 2 |
| Ferroptotic cell death releases DAMPs (damage‐associated molecular patterns), which stimulate microglial activation and inflammatory cytokine release (e.g., IL‐1β, TNF‐α), aggravating neuroinflammation and secondary neuronal injury post‐stroke. | Establishes ferroptosis as a contributor to both primary neuronal death and post‐ischemic inflammatory cascades. | He et al. ( |
| 3 |
| Novel CSF and plasma biomarkers such as oxylipins (oxidized lipid mediators), GPX4 fragments, and 8‐iso‐prostaglandin F2α have been identified as indicators of ferroptosis activity and oxidative lipid damage in stroke patients. | Paves the way for developing noninvasive, real‐time diagnostic tools for early detection and monitoring of ferroptosis. | Liu et al. ( |
| 4 |
| Nanoparticle formulations (e.g., liproxstatin‐1‐loaded liposomes or polymeric carriers) have demonstrated enhanced BBB penetration, controlled drug release, and improved localization in infarcted brain regions. | Offers a promising strategy for targeted ferroptosis inhibition with improved pharmacokinetics and neuroprotection. | Shi et al. ( |
| 5 |
| A first‐in‐human pilot trial (NCT05938447) is testing deferoxamine, an iron chelator, in patients with acute ischemic stroke and elevated serum iron. The goal is to evaluate safety, feasibility, and potential neuroprotective effects via ferroptosis modulation. | Marks early clinical translation of ferroptosis‐targeted therapy, bridging bench‐to‐bedside application. | Millán et al. ( |
| Focus area | What has been achieved | What current research is exploring | References |
|---|---|---|---|
|
| Confirmed mitochondrial ROS and iron cluster disruption as triggers of ferroptosis | Development of mitochondrial‐targeted antioxidants to mitigate ferroptotic signaling | Feng et al. ( |
|
| Demonstrated DAMP‐mediated cytokine release post‐ferroptosis | Investigating dual inhibitors targeting ferroptosis and inflammation (e.g., Nrf2‐NF‐κB axis modulators) | Yu et al. ( |
|
| Identified oxylipins, GPX4 fragments, 8‐iso‐PGF2α as potential markers | Validation in human cohorts; integration into clinical diagnostic panels | van ’t Erve et al. ( |
|
| Liproxstatin‐1 nanoparticles improved BBB delivery and neuroprotection | Optimizing ligand‐targeted nanocarriers for precise ischemic tissue localization | Renshuai et al. ( |
| Combination regimen | Mechanistic synergy | Experimental evidence/model outcomes | Key primary references |
|---|---|---|---|
|
| Fer‐1 scavenges lipid peroxyl radicals and prevents ferroptotic membrane damage; tPA restores cerebral perfusion. Combined use reduces I/R‐induced oxidative burst. | In MCAO models, Fer‐1 coadministration reduces infarct size and neuronal lipid peroxidation during post‐tPA reperfusion without impairing thrombolysis. | Entezari et al. |
|
| DFO chelates Fe2 + and suppresses Fenton chemistry; minocycline inhibits microglial activation and downstream inflammatory cytokines. | Combined therapy reduces iron accumulation, microglial polarization, and neuronal ferroptosis in I/R models. | Briones‐Valdivieso et al. ( |
|
| Sulforaphane activates Nrf2, enhancing GPX4, HO‐1, and GSH synthesis; edaravone scavenges ROS and inhibits lipid peroxidation. | Dual treatment enhances antioxidant capacity, reduces mitochondrial ROS, and improves neurological recovery. | Hu et al. |
|
| Lip‐1 inhibits lipid peroxidation; TNF‐α blockade reduces neuroinflammation and suppresses cytokine‐driven ferroptosis amplification. | Demonstrated reduction of BBB disruption, cytokine burden, and ferroptotic neuron loss in rodent MCAO models. | Huang et al. |
| S. No. | Compound | Source | Mechanism of action | Evidence in cerebral infarction | References |
|---|---|---|---|---|---|
| 1. | Quercetin | Flavonoid | Antioxidant, inhibits lipid peroxidation, modulates Nrf2/GPX4 | Shown to reduce ferroptosis and protect neurons in stroke models | Zheng et al. ( |
| 2. | Baicalin/baicalein | Flavonoid (Scutellaria baicalensis) | Activates Nrf2/ARE, upregulates GPX4, reduces iron overload | Demonstrated neuroprotection in ischemic stroke | Li et al. ( |
| 3. | Epigallocatechin‐3‐gallate (EGCG) | Green tea polyphenol | Antioxidant, inhibits lipid peroxidation | Shown to inhibit ferroptosis in CNS models | Zheng et al. ( |
| 4. | Berberine | Alkaloid (Coptis chinensis) | Modulates iron homeostasis, antioxidant | Shown to protect against ischemic injury | Bi‐Tiao et al. ( |
| 5. | Salvianolic acid A | Polyphenol (Salvia miltiorrhiza) | Iron chelator, inhibits lipid peroxidation | Reduces neuronal ferroptosis in hemorrhagic stroke | Shi et al. ( |
| 6. | Icariside II | Flavonoid (Epimedium) | Activates Nrf2/GPX4, reduces oxidative stress | Shown to inhibit ferroptosis after ischemia | Zhang et al. ( |
| 7. | Dl‐3‐n‐butylphthalide | Synthetic from celery seed | Antioxidant, modulates iron metabolism | Effective in animal stroke models | Xu et al. ( |
| 8. | Ginkgolide B | Terpene lactone (Ginkgo biloba) | Antioxidant, anti‐inflammatory | Demonstrated efficacy in stroke models | Xu et al. ( |
| 9. | Loureirin C | Flavonoid (Dragon's blood) | Antioxidant, modulates iron metabolism | Potential neuroprotective effects | Xu et al. ( |
| 10. | Astragaloside IV | Saponin (Astragalus membranaceus) | Inhibits ferroptosis‐associated pathways | Shown to alleviate brain injury in stroke | Zhang et al. ( |
| 11. | Resveratrol | Polyphenol (grapes, berries) | Inhibits ferroptosis‐induced cell death | Shown to reduce ischemia/reperfusion injury | Li et al. ( |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsFerroptosis and cancer prognosis · Clusterin in disease pathology · Cancer-related molecular mechanisms research
Introduction
1
Cerebral infarction, commonly referred to as ischemic stroke (IS), is a major cause of mortality and long‐term disability worldwide. It occurs when a cerebral artery is obstructed, leading to the deprivation of oxygen and nutrients in brain tissue, ultimately resulting in neuronal death and loss of neurological function. The pathological cascade following cerebral infarction involves a complex interplay of excitotoxicity, oxidative stress, inflammation, and various forms of cell death (Kuriakose and Xiao 2020). While traditional forms of cell death, such as necrosis and apoptosis, have long been studied in the context of cerebral infarction, recent research has brought attention to a novel form of regulated cell death, ferroptosis, as a key contributor to neuronal damage in IS (Salaudeen et al. 2024). Ferroptosis is an iron‐dependent, nonapoptotic form of cell death characterized by the accumulation of lipid peroxides and reactive oxygen species (ROS) (Chai et al. 2024). Unlike apoptosis or necrosis, ferroptosis is driven by iron‐catalyzed oxidative damage to cellular membranes, particularly phospholipids containing polyunsaturated fatty acids (PUFAs) (Zhang et al. 2022). Central to this process is the enzyme glutathione peroxidase 4 (GPX4), which normally protects cells by reducing lipid hydroperoxides to nontoxic lipid alcohols (Weaver and Skouta 2022; Manikandan et al. 2025). Inhibition of GPX4, depletion of glutathione (GSH), or disruption of cystine uptake through System Xc^−^ (Cystine/Glutamate Antiporter System) impairs the cell's antioxidant defenses, promoting uncontrolled lipid peroxidation and triggering ferroptosis (Li et al. 2022). The identification of ferroptosis as a critical mediator of neuronal injury in cerebral infarction represents a paradigm shift in stroke research, offering novel insights into pathophysiological mechanisms and opening new avenues for targeted therapeutic interventions (Jing et al. 2024).
Leads to the cerebral infarction, ischemia−reperfusion (I/R) injury primes a surge in oxidative stress and iron overload, creating a favorable environment for ferroptosis. Accumulating evidence from in vivo and in vitro studies has shown elevated markers of ferroptosis in ischemic brain regions, including increased iron accumulation, reduced GPX4 expression, and elevated levels of malondialdehyde (MDA) and 4‐hydroxynonenal (4‐HNE), hallmarks of lipid peroxidation (Lillo‐Moya et al. 2021). Furthermore, pharmacological inhibition of ferroptosis using agents like ferrostatin‐1, liproxstatin‐1, and iron chelators such as deferoxamine (DFO) has been shown to reduce infarct size, attenuate neuronal death, and improve functional recovery in experimental stroke models (Kailin et al. 2022). Recent advances in this field have also identified novel regulatory pathways and molecules involved in ferroptosis, such as the Nrf2 (nuclear factor erythroid 2–2‐related factor 2) antioxidant response, FSP1‐CoQ10 axis, and lipid metabolic enzymes like acyl‐CoA synthetase long‐chain family member 4 (ACSL4) and lysophosphatidylcholine acyltransferase 3 (LPCAT3), offering new insights into potential therapeutic targets (Tang et al. 2021; Yan et al. 2023). Moreover, the discovery of GPX4‐independent ferroptosis‐suppressing mechanisms expands the therapeutic landscape and underlines the complexity of redox regulation in the ischemic brain (Li et al. 2022). However, in this review, we will discuss the ferroptosis molecular mechanism and how it is involved in cerebral infarction. As well as ongoing investigations and progress into the molecular regulation and modulation of ferroptosis continue to advance our understanding of its role in ischemic brain injury, highlighting the need for translational strategies that can effectively harness this knowledge, thereby marking significant research progress in the field of ferroptosis in cerebral infarction.
Pathophysiology of Ferroptosis in Cerebral Infarction
2
Following the onset of cerebral ischemia and during reperfusion, the brain experiences a dramatic increase in oxidative stress, marked by the excessive generation of ROS. This oxidative burden overwhelms endogenous antioxidant defense systems and initiates lipid peroxidation, a key biochemical hallmark of ferroptosis (Salaudeen et al. 2024). At the same time, ischemic conditions disrupt iron homeostasis, leading to iron accumulation in neuronal tissues. In particular, excess ferrous iron (Fe^2^ ^+^) catalyzes the Fenton reaction, producing hydroxyl radicals (OH) that intensify oxidative damage by initiating the peroxidation of PUFA‐containing phospholipids in neuronal membranes (Fardin and Ali 2025). Central to ferroptosis regulation is GPX4, a lipid hydroperoxide detoxifying enzyme that uses GSH as a cofactor to convert toxic lipid peroxides into inert lipid alcohols (Long et al. 2023). During cerebral infarction, GPX4 activity is compromised due to intracellular GSH depletion and impaired cystine uptake. The latter is mediated by dysfunction of the System Xc^−^ transporter, which under normal physiological conditions imports cystine in exchange for glutamate. Under ischemic conditions, elevated extracellular glutamate concentrations caused by excitotoxicity inhibit System Xc^−^, further reducing cystine uptake, impairing GSH biosynthesis, and weakening the cell's antioxidant defense (Feng et al. 2023). Moreover, lipid metabolism enzymes such as ACSL4 and LPCAT3 play a significant role in incorporating PUFAs into phospholipids, making them prime substrates for peroxidation. The accumulation and oxidation of these lipid species compromise membrane integrity, resulting in iron‐dependent, nonapoptotic neuronal death, characteristic of ferroptosis (Sun et al. 2025). The mechanism and pathway of ferroptosis in cerebral infarction are shown in Figure 1.
Mechanistic pathways underlying ferroptosis in cerebral infarction. This schematic illustrates the major molecular events that drive ferroptotic neuronal death following cerebral ischemia and reperfusion. Disruption of iron homeostasis increases intracellular Fe2 +, which participates in the Fenton reaction to generate hydroxyl radicals (·OH) and amplify oxidative stress. In parallel, mitochondrial dysfunction during ischemia–reperfusion promotes excessive production of reactive oxygen species and destabilizes iron–sulfur clusters, further enriching the labile iron pool. Lipid peroxidation is initiated when ACSL4 and LPCAT3 incorporate polyunsaturated fatty acids (PUFAs) into membrane phospholipids, creating substrates for oxidation by lipoxygenases (LOX). Accumulating lipid hydroperoxides (LOOH) compromise membrane integrity and act as central executors of ferroptosis. Under physiological conditions, the System Xc− transporter supplies cystine for glutathione (GSH) synthesis, enabling glutathione peroxidase‐4 (GPX4) to reduce LOOH to nontoxic lipid alcohols. During cerebral infarction, excitotoxic glutamate elevation and oxidative depletion of GSH inhibit the System Xc−/GSH/GPX4 axis, rendering neurons vulnerable to unrestricted lipid peroxidation. Ferroptotic cell death also interfaces with neuroinflammation: the release of damage‐associated molecular patterns (DAMPs) activates microglia, leading to cytokine production (e.g., IL‐1β, TNF‐α) that reinforces oxidative injury and ferroptosis. Together, these converging pathways iron dysregulation, mitochondrial ROS, impaired antioxidant defense, and inflammatory amplification drive ferroptosis as a key mechanism of neuronal injury in cerebral infarction.
Collectively, these converging disturbances in iron handling, redox balance, lipid metabolism, and antioxidant defense establish a cellular environment that strongly predisposes neurons to ferroptotic injury during cerebral infarction. The mechanistic framework outlined above underscores the centrality of ferroptosis within the broader I/R cascade. To substantiate these molecular pathways, a growing body of in vivo and in vitro research has provided direct experimental evidence demonstrating the occurrence and functional relevance of ferroptosis in stroke models.
Experimental Evidence Supporting Ferroptosis in Stroke Models
2.1
Building on this mechanistic foundation, experimental studies in established IS models—particularly the middle cerebral artery occlusion (MCAO) model—have validated the biochemical and morphological hallmarks of ferroptosis and clarified its contribution to neuronal loss after ischemic injury. Preclinical studies using animal models of cerebral ischemia, particularly the MCAO model, have provided compelling evidence for the role of ferroptosis in stroke pathology. These studies report increased levels of lipid peroxidation markers, such as MDA and 4‐HNE, along with suppressed GPX4 expression and elevated iron deposition in the ischemic brain (Tuo and Lei 2024). Crucially, pharmacological inhibition of ferroptosis using agents like ferrostatin‐1, liproxstatin‐1, and DFO (an iron chelator) has been shown to reduce infarct volume, preserve neuronal integrity, and improve post‐stroke neurological function (Youjun et al. 2025).
Ferroptosis and Secondary Injury Mechanisms
2.2
Beyond its role in initiating neuronal death, ferroptosis significantly contributes to secondary injury mechanisms. Ferroptotic neurons release damage‐associated molecular patterns (DAMPs), which activate microglia and astrocytes, leading to elevated production of proinflammatory cytokines (e.g., IL‐1β, TNF‐α) and the amplification of neuroinflammation (Kang et al. 2024). Additionally, ferroptosis exacerbates BBB disruption, facilitating the infiltration of peripheral immune cells and further promoting inflammatory damage (Xu et al. 2024). Mitochondrial dysfunction, another hallmark of I/R injury, adds to this cascade by generating excessive mitochondrial ROS and contributing to the disruption of iron–sulfur clusters, which accelerates ferroptosis progression (Xing‐Yu et al. 2024). Recent research has broadened our understanding of ferroptosis‐related mechanisms in cerebral infarction, including mitochondrial involvement, immune crosstalk, and translational biomarkers. These advancements are summarized in Table 1.
Complementing these advances, early phase clinical trials have begun exploring the safety and feasibility of ferroptosis modulators like DFO in acute IS patients with iron overload, marking a critical step toward clinical translation. Together, these findings underscore the expanding recognition of ferroptosis not only as a pathological hallmark but also as a modifiable and actionable target in cerebral infarction. Advances in mechanistic insight, biomarker discovery, targeted drug delivery, and therapeutic evaluation are collectively propelling this field toward innovative, ferroptosis‐centered approaches for stroke treatment and recovery. Collectively, these insights underscore the central role of ferroptosis in the pathophysiology of cerebral infarction. The convergence of oxidative stress, iron dysregulation, impaired redox homeostasis, and excitotoxicity drives ferroptotic neuronal death and secondary brain injury. Advances in mechanistic elucidation, biomarker discovery, drug delivery systems, and clinical testing have laid a robust foundation for translating ferroptosis‐targeted strategies into future therapeutic interventions (Liu et al. 2024). As the field continues to evolve, integrating ferroptosis modulation into stroke management may offer new hope for reducing morbidity and enhancing recovery in IS patients.
Mechanistic Integration, Research Progress, and Current Directions
3
The pathogenesis of ferroptosis in cerebral infarction is underpinned by several interconnected molecular mechanisms that reflect the complex biology of ischemic neuronal death. Emerging evidence has begun to elucidate how these mechanisms, namely, mitochondrial dysfunction, oxidative stress, neuroinflammation, iron metabolism, and impaired antioxidant defense, collectively orchestrate ferroptosis, offering novel avenues for targeted therapeutic intervention.
Mitochondrial Dysfunction as a Ferroptosis Amplifier
3.1
Mitochondria play a central role in ferroptosis by regulating redox balance, energy metabolism, and iron homeostasis. Under ischemic stress, mitochondrial respiration becomes impaired, leading to electron leakage at complexes I and III of the electron transport chain. This leakage produces excessive mitochondrial ROS, especially superoxide (O_2_ ^−^), which can be further converted into hydroxyl radicals via the Fenton reaction, exacerbated by iron overload (Tian et al. 2023). Moreover, iron–sulfur (Fe–S) cluster‐containing enzymes in the mitochondrial matrix, such as aconitase and complex I, undergo disintegration during oxidative stress, releasing redox‐active Fe^2^ ^+^ ions. This labile iron accelerates lipid peroxidation in mitochondrial membranes enriched in cardiolipin and other polyunsaturated phospholipids, contributing to ferroptotic signaling (Feng et al. 2024). Recent findings confirm that I/R disrupts mitochondrial morphology (e.g., outer membrane rupture, cristae loss), increases mitochondrial iron content, and reduces membrane potential hallmarks of ferroptosis. Current investigations are evaluating mitochondria‐targeted antioxidants like MitoQ, SkQ1, and SS‐31, along with iron chelators localized to mitochondria, to mitigate ferroptotic injury at its source (Feng et al. 2023; Salis Torres et al. 2024).
Neuroinflammation–Ferroptosis Crosstalk
3.2
Ferroptosis does not occur in isolation but intersects with the inflammatory landscape of the ischemic brain. Ferroptotic neurons release DAMPs, oxidized phospholipids, and lipid peroxidation products such as 4‐HNE and MDA. These molecules are sensed by pattern recognition receptors like TLR4 on microglia and astrocytes, triggering proinflammatory signaling cascades. Activated glial cells release cytokines such as IL‐1β, IL‐6, TNF‐α, and chemokines, contributing to secondary neuronal damage (Guo et al. 2025; Chen et al. 2024). Recent research contributes to potential therapeutic roles of various natural products in regulating neuroinflammation and ferroptosis. The results suggest that natural products have significant therapeutic potential in modulating the interaction between neuroinflammation and ferroptosis, making them potential treatments for Parkinson's disease (PD) (Guo et al. 2025). Future research should further validate the safety and efficacy of these natural compounds in clinical applications to develop novel therapeutic strategies for PD (Xu et al. 2024). Notably, this inflammatory microenvironment also feeds back into ferroptosis by increasing ROS production, inhibiting System Xc^−^, and reducing GPX4 expression. This bidirectional loop amplifies brain injury after stroke. Recent research demonstrated that blocking NF‐κB signaling attenuates both inflammation and ferroptotic neuronal death in ischemic models. In addition, although studies have shown that ferroptosis incidence is highly correlated with LPO, only a small number of ferroptotic inhibitors target lipid‐rich mitochondria and cytomembrane with weak specificity. In the traditional sense, selenocysteine active sites in GPX4 and FSP‐1 play decisive roles in resisting ferroptosis. Therapeutically, dual‐pathway inhibitors targeting Nrf2 activation and NF‐κB suppression are under exploration to disrupt this pathogenic interface (Liu et al. 2024).
Biomarker Discovery: From Bench to Bedside
3.3
Recent research focuses on identifying biomarkers associated with ferroptosis to improve diagnosis, prognosis, and therapeutic targeting in cerebral infarction.
Key Research Studies
- Gene Pair Biomarkers in IS
- Study Focus: Identification of ferroptosis‐related gene pair biomarkers in IS and their roles in immune infiltration.
- Findings: The gene pair CDKN1A/JUN was identified as a robust diagnostic biomarker, outperforming single‐gene markers. These genes are implicated in regulating ferroptosis during IS progression and are linked to immune cell infiltration, particularly plasma cells (Fan et al. 2022).
- Nfe2l2 as a Diagnostic Biomarker
- Study Focus: Investigation of ferroptosis‐related biomarkers in IS and their effects on immune infiltration using single‐cell RNA sequencing and animal models.
- Findings: Nfe2l2 (nuclear factor erythroid‐derived 2‐like 2) was identified as a potential diagnostic biomarker. The study suggests that targeting the Nfe2l2/HO‐1 pathway may suppress ferroptosis and offer therapeutic benefits in IS (Fan et al. 2025).
- Oxidative Stress‐Ferroptosis Biomarkers
- Study Focus: Analysis of oxidative stress and ferroptosis‐related genes in stroke.
- Findings: Four biomarkers, CDKN1A, GPX4, PRDX1, and PRDX6, were highlighted for diagnosing stroke and assessing treatment effects. These genes are involved in oxidative stress and ferroptosis pathways (Zhang et al. 2024).
- PTGS2 as a Diagnostic Marker
- Study Focus: Identification of diagnostic biomarkers at the intersection of oxidative stress and ferroptosis.
- Findings: PTGS2 was found to be a differentially expressed gene of ferroptosis in IS. Inhibition of PTGS2 improved neurological outcomes and reduced infarct volume in animal models, suggesting its potential as a therapeutic target (Yuan et al. 2024).
- Mechanistic Reviews and Additional Biomarkers
- Mechanisms: Reviews have summarized the role of iron metabolism (e.g., serum ferritin, transferrin, hepcidin), glutamate metabolism, and antioxidant systems (e.g., GPX4, GSH, Nrf2 pathway) in ferroptosis during IS.
- Clinical Relevance: Biomarkers such as serum ferritin, transferrin, hepcidin, and ceruloplasmin have been associated with stroke prognosis and ferroptosis activity in clinical and experimental settings (Deng et al. 2023).
Nanocarrier‐Based Therapeutic Delivery
3.4
- ROS‐Responsive Nanoparticles: Recent studies have developed ROS‐responsive nanoparticles that deliver ferroptosis inhibitors in a controlled manner. For example, a system combining human umbilical cord mesenchymal stem cells (Huc‐MSCs) and a ferroptosis inhibitor demonstrated synergistic neuroprotection in ischemic models by anchoring the therapeutic payload at the site of injury and releasing it in response to oxidative stress (Renshuai et al. 2024).
- Iron Oxide Nanoparticles (IONPs): IONPs have been explored for their ability to induce ferroptosis in cancer, but in the context of stroke, their application is more nuanced, acting as either imaging agents or, when combined with antioxidants/ferroptosis inhibitors, as neuroprotective carriers (Sant'Angelo et al. 2025).
- Functionalized Nanoparticles: Reviews highlight that functionalized nanoparticles (e.g., those decorated with targeting ligands) can cross the BBB and deliver ferroptosis modulators directly to ischemic brain tissue, enhancing efficacy and reducing systemic toxicity (Ziying et al. 2024).
- Natural Product‐Loaded Nanoparticles: Some natural antioxidants and ferroptosis inhibitors with poor bioavailability (such as N‐acetylcysteine and its derivatives) are being encapsulated in nanoparticles to improve brain delivery and therapeutic effect (Sun et al. 2023).
Nanoparticle‐based delivery systems, including ROS‐responsive, functionalized, and biomimetic nanoparticles, are at the forefront of preclinical research for targeting ferroptosis in cerebral infarction. These systems enable precise, BBB‐penetrant, and stimuli‐responsive delivery of ferroptosis inhibitors, iron chelators, and antioxidants, demonstrating significant neuroprotection in animal models. The field is rapidly evolving, with ongoing studies optimizing delivery vehicles and targeting strategies for future clinical translation (Sun et al. 2023).
Clinical Translation and Ongoing Trials
3.5
There are no registered or published clinical trials to date that specifically target ferroptosis in cerebral infarction (IS) in human patients (Dong et al. 2024; Maremonti et al. 2024). The field is still largely in the preclinical and translational research stage. Recent high‐impact studies, such as the 2025 investigation of the ATM inhibitor AZD1390, have demonstrated in vitro, ex vivo, and in vivo (animal) efficacy in reducing ferroptosis and protecting brain tissue after IS. However, these studies have not yet advanced to human clinical trials (Lee et al. 2025). Reviews from 2024 and 2025 emphasize the therapeutic potential of ferroptosis inhibitors (such as Fer‐1, DFO, and GPX4 activators) in stroke models, but also highlight that the lack of validated biomarkers for ferroptosis in humans and insufficient clinical data currently impede clinical trial development. Some iron chelators and antioxidants have been clinically tested for stroke, but not specifically as ferroptosis‐targeted therapies. As outlined in Table 2, ongoing efforts are focused on bridging this gap by refining biomarkers, improving delivery systems, and developing targeted interventions to accelerate the clinical translation of ferroptosis‐based therapies.
The expanding understanding of ferroptosis in IS has revealed a multidimensional network of mechanisms involving mitochondria, inflammatory signaling, redox imbalance, and iron metabolism. Recent progress in biomarker discovery, nanocarrier systems, and early phase clinical trials highlights the translational momentum of this field (Gowtham et al. 2024). Future research must now focus on integrating multitarget strategies, refining diagnostic tools, and accelerating the clinical application of ferroptosis‐targeted therapies to enhance stroke outcomes (Bu et al. 2021).
Therapeutic Strategies Targeting Ferroptosis in Cerebral Infarction
4
Recent advancements in understanding the molecular mechanisms of ferroptosis have catalyzed the development of targeted interventions aiming to mitigate ferroptosis‐induced neuronal death in IS. These strategies span from direct inhibition of lipid peroxidation to modulation of redox balance, iron metabolism, and gene expression (Chen et al. 2024). Below, we discuss the main therapeutic avenues currently under investigation.
Pharmacological Inhibitors of Ferroptosis
4.1
The most direct approach to mitigating ferroptosis is through small‐molecule inhibitors that interfere with its core molecular drivers: lipid peroxidation, iron accumulation, and GSH system failure.
Lipid Peroxidation Inhibitors
4.1.1
Small molecules like ferrostatin‐1 (Fer‐1) and liproxstatin‐1 (Lip‐1) act as lipophilic radical‐trapping antioxidants, scavenging lipid peroxyl radicals (LOO•) and halting the peroxidation chain reaction. These agents preserve membrane integrity and mitochondrial function in rodent models of stroke, especially in MCAO and global ischemia models (Huang et al. 2023).
GPX4 Modulators
4.1.2
GPX4 is a central enzyme that reduces lipid hydroperoxides (LOOH) to nontoxic lipid alcohols using GSH. Agents that upregulate GPX4, such as baicalein and selenium supplementation, enhance cellular defense mechanisms against ferroptosis. Moreover, targeting upstream molecules like SLC7A11, which imports cystine for GSH synthesis, offers indirect protection (Hu et al. 2019).
Iron Chelators
4.1.3
Iron chelators such as:
- DFO: An FDA‐approved agent for iron overload, now in stroke trials.
- Deferiprone and deferasirox: Oral iron chelators with potential Central Nervous System (CNS) applications.
- VK‐28 and HBED: Brain‐penetrant experimental chelators (Mobarra et al. 2016; Entezari et al. 2022).
These chelators attenuate iron‐mediated ROS generation and disrupt the Fenton reaction (Fe^2^ ^+^ + H_2_O_2_ → Fe^3^ ^+^ + OH + OH^−^), a core contributor to ferroptosis initiation (Entezari et al. 2022).
Nrf2 Pathway Activators
4.1.4
The Nrf2 pathway orchestrates the cellular antioxidant response. Activators like sulforaphane, dimethyl fumarate, and bardoxolone methyl promote the transcription of antioxidant genes, including SLC7A11, GPX4, HO‐1, and NQO1, restoring redox balance and ferroptosis resistance (Chen and Maltagliati 2018).
Combination Therapies: A Multitargeted Approach
4.2
Cerebral infarction is characterized by a complex interplay of oxidative stress, excitotoxicity, inflammation, mitochondrial dysfunction, and multiple forms of regulated cell death, including ferroptosis. Because these mechanisms are temporally and spatially interconnected, single‐target interventions often provide only partial neuroprotection. Recent preclinical studies increasingly support the use of combination therapies designed to modulate several pathogenic pathways simultaneously (Entezari et al. 2022; Chen and Maltagliati 2018). Such multimodal strategies are particularly relevant in I/R injury, where the abrupt restoration of blood flow triggers a surge in ROS, iron mobilization, lipid peroxidation, and inflammatory signaling conditions that strongly potentiate ferroptosis. Combination regimens pairing ferroptosis inhibitors with thrombolytics, anti‐inflammatory agents, iron chelators, or antioxidant compounds have shown synergistic benefits in experimental stroke models. For example, coadministration of radical‐trapping antioxidants with recombinant tissue plasminogen activator reduces I/R‐induced oxidative and lipid peroxidation damage without compromising thrombolytic efficacy (Huang et al. 2023; Mobarra et al. 2016). Similarly, iron chelation combined with microglial‐targeted agents attenuates both ferroptotic injury and inflammatory cascades. Other combinations, such as Nrf2 activators with ROS scavengers, enhance endogenous antioxidant defenses, whereas ferroptosis inhibitors paired with cytokine blockers mitigate secondary neuroinflammatory injury. Collectively, these emerging strategies highlight the therapeutic promise of coordinated, multitarget modulation of ferroptosis, inflammation, iron metabolism, and oxidative injury. Translating such regimens to clinical practice will require optimization of dosing windows, pharmacokinetics, and interactions with standard‐of‐care reperfusion therapies (Entezari et al. 2022; Briones‐Valdivieso et al. 2024). A summary of the representative combination has been shown in Table 3.
Natural Compounds and Nutraceuticals
4.3
Natural compounds have demonstrated significant potential in modulating ferroptosis‐related pathways, offering promising therapeutic strategies for conditions like cerebral infarction. One of the key mechanisms involves activation of the Nrf2/ARE pathway, where several flavonoids such as quercetin, baicalin, and kaempferol enhance antioxidant defenses by upregulating genes like GPX4, a crucial enzyme that suppresses lipid peroxidation (Li et al. 2025; Shi et al. 2024). Additionally, iron chelation has emerged as a vital approach, with compounds like salvianolic acid A and the clinically used DFO effectively reducing iron overload, a central trigger of ferroptosis (Shi et al. 2024). Natural antioxidants such as baicalein and epigallocatechin gallate also play a pivotal role by directly scavenging ROS and inhibiting the chain reaction of lipid peroxidation, thereby preserving neuronal membrane integrity (Bi‐Tiao et al. 2025). Traditional Chinese Medicine and herbal formulations further contribute to ferroptosis regulation. For example, Danhong injection, comprising Salvia miltiorrhiza and saffron, has shown efficacy in improving outcomes in IS by modulating ferroptotic processes (Chen et al. 2024). Moreover, nonpharmacological interventions like moxibustion have demonstrated neuroprotective effects through the inhibition of ROS accumulation and ferroptotic neuronal death. Collectively, these natural approaches represent a multifaceted strategy for targeting ferroptosis in neurological disorders. A summary of these compounds and their specific mechanisms of action is provided in Table 4, offering a concise overview of their therapeutic relevance.
These compounds are being tested in various animal models and may serve as long‐term maintenance therapy for high‐risk patients or stroke survivors to prevent secondary injury. These agents act through antioxidant, iron‐chelating, and antilipid peroxidation mechanisms, often modulating key signaling pathways such as Nrf2/GPX4. While preclinical data are encouraging, further clinical research is needed to translate these findings into effective treatments for stroke patients.
Gene and RNA‐Based Therapeutic Strategies
4.4
Targeting ferroptosis at the genetic and transcriptional level provides a more specific and durable strategy to modulate neuronal vulnerability.
RNA‐Based Therapeutic Approaches
4.4.1
RNA‐based therapeutic strategies are gaining momentum as innovative approaches to modulate ferroptosis, particularly in the context of neurodegenerative and ischemic conditions such as stroke (Henninger and Mayasi 2019). Noncoding RNAs (ncRNAs), including microRNAs (miRNAs), long noncoding RNAs (lncRNAs), and circular RNAs (circRNAs), have emerged as key regulators of ferroptosis through their control over gene expression and signaling pathways (Wu et al. 2025; Zeyu et al. 2022). Among miRNAs, miR‐194 enhances cell survival and suppresses ferroptosis by targeting the repressor Bach1, thereby activating the Nrf2/HO‐1 antioxidant pathway. Similarly, miR‐210‐3p, when delivered via hypoxia‐conditioned exosomes, downregulates transferrin receptor expression, effectively limiting iron uptake and reducing ferroptosis. Another miRNA, miR‐23a‐3p, carried by mesenchymal stem cell (MSC)‐derived exosomes, mitigates ferroptotic damage by lowering oxidative stress and ferroptosis biomarkers (Wei et al. 2022). In terms of lncRNAs, Mir9‐3hg identified in bone marrow MSC‐derived exosomes suppresses ferroptosis by modulating RNA‐binding proteins and antioxidant enzyme expression. circRNAs, though still under active investigation, have shown dual roles, either promoting or inhibiting ferroptosis, suggesting their potential both as diagnostic biomarkers and therapeutic targets. Notably, the exosomal delivery of these ncRNAs from stem cells has shown substantial promise in preclinical models by attenuating ferroptosis and ameliorating ischemic injury. These findings underscore the therapeutic relevance of ncRNAs and pave the way for RNA‐based interventions in ferroptosis‐related neurological disorders (Zhang and Xie 2024).
CRISPR‐Cas9 Gene Editing
4.4.2
Permanent disruption or repair of ferroptosis‐related genes (e.g., GPX4, SLC7A11, FSP1) using CRISPR could offer long‐term neuroprotection, especially in genetically predisposed individuals. This requires caution due to off‐target risks but holds immense promise. One landmark study used a CRISPR–Cas9 screening strategy to identify FSP1 (ferroptosis suppressor protein 1) as a potent inhibitor of ferroptosis. FSP1 was validated as a key target in both hemorrhagic and IS models, suggesting that gene editing of FSP1 or related pathways could modulate ferroptosis and influence stroke outcomes (Xu et al. 2023).
Antisense Oligonucleotides
4.4.3
Antisense oligonucleotides (ASOs) can downregulate ferroptosis drivers such as p53, which suppresses SLC7A11, and NCOA4, which promotes ferritinophagy (iron release from ferritin). Early preclinical studies show positive outcomes (Liu and Gu 2022). A relevant study discusses the potential of ASOs to downregulate key ferroptosis drivers such as p53 and NCOA4, which are involved in promoting ferroptosis through suppression of SLC7A11 and ferritinophagy (iron release), respectively. Early preclinical evidence suggests that targeting these ferroptosis regulators with ASOs can mitigate neuronal death and cerebral ischemic injury by reducing iron overload and lipid peroxidation in stroke models (Xu et al. 2023).
Clinical Translation: Progress and Roadblocks
4.5
While preclinical data are encouraging, clinical translation remains in early phases. The first clinical trial (NCT05938447) is testing DFO in patients with acute IS and elevated iron markers. Initial goals are to evaluate safety, pharmacodynamics, and preliminary efficacy.
Translational Challenges Include:
- Therapeutic window: Ferroptosis inhibitors must be administered within hours of stroke onset.
- Patient variability: Stroke subtypes (lacunar, embolic, hemorrhagic) may respond differently.
- Lack of real‐time biomarkers: Hinders the identification of ferroptosis‐active patients.
- Systemic side effects: Especially with iron chelators or GPX4 modulators.
Future Directions:
- Personalized medicine: Tailoring treatments based on iron load, oxidative stress markers, and genomics.
- Advanced neuroimaging: Use of iron‐sensitive MRI, diffusion tensor imaging, and MRS to guide therapy.
- Larger phase II/III trials: Needed to validate efficacy and define clinical endpoints.
Therapeutic targeting of ferroptosis in cerebral infarction is advancing rapidly, integrating insights from redox biology, iron metabolism, and molecular pharmacology. The combination of traditional small‐molecule drugs, natural antioxidants, gene‐editing technologies, and nanocarrier systems offers a multidimensional arsenal against ischemic brain injury. As research transitions from bench to bedside, strategic efforts must prioritize clinical validation, patient stratification, and timely intervention to harness the full therapeutic potential of ferroptosis modulation.
Conclusion
5
Ferroptosis has emerged as a critical and distinct form of regulated cell death in the pathophysiology of cerebral infarction, offering a paradigm shift in our understanding of ischemic neuronal injury. Unlike necrosis or apoptosis, ferroptosis is characterized by iron‐dependent lipid peroxidation, compromised antioxidant defenses, particularly GPX4 inactivation and GSH depletion, and mitochondrial oxidative stress. These molecular hallmarks of ferroptosis are increasingly recognized as central contributors not only to primary neuronal death but also to secondary injury mechanisms, including blood–brain barrier disruption, neuroinflammation, and mitochondrial dysfunction in the ischemic brain.
Over the past decade, substantial progress has been made in dissecting the molecular architecture of ferroptosis in cerebral ischemia. Studies have delineated the roles of key players such as System Xc^−^, ACSL4, Nrf2, and iron‐regulatory proteins like ferritin, TfR1, and DMT1 in driving ferroptotic cascades. Moreover, mitochondrial ROS generation, iron–sulfur cluster instability, and ferritinophagy have been identified as amplifiers of ferroptotic injury in neurons and vascular endothelial cells. Importantly, these findings underscore that ferroptosis is not an isolated process but rather a converging node where redox imbalance, iron overload, excitotoxicity, and inflammation intersect.
From a translational standpoint, preclinical evidence is robust and promising. Pharmacological inhibition of ferroptosis through lipid ROS scavengers (ferrostatin‐1, liproxstatin‐1), iron chelators (DFO), GPX4 stabilizers, and Nrf2 activators has demonstrated consistent neuroprotective effects in rodent models of MCAO. These interventions have shown benefits such as reduced infarct volume, preserved neuronal integrity, improved behavioral outcomes, and delayed neurodegeneration.
Recent innovations such as nanocarrier‐based delivery systems, gene and RNA‐based therapies, and biomarker discovery are accelerating the transition of ferroptosis research from bench to bedside. The initiation of early phase human trials evaluating DFO in acute IS (NCT05938447) signals an encouraging move toward clinical translation. However, challenges remain in terms of drug delivery across the blood–brain barrier, defining optimal therapeutic windows, and stratifying patients based on ferroptosis susceptibility using biomarkers or imaging tools.
Looking ahead, future research should focus on:
- Clarifying the temporal dynamics of ferroptosis during the acute, subacute, and chronic phases of stroke.
- Integrating ferroptosis modulation with standard stroke therapies such as thrombolytics and neurorehabilitation.
- Personalizing interventions based on age, sex, iron status, and genetic background.
- Exploring ferroptosis‐inflammation crosstalk as a dual therapeutic target.
- Developing real‐time diagnostic tools to monitor ferroptosis in vivo.
In conclusion, ferroptosis represents not only a novel and actionable mechanism of neuronal death in cerebral infarction but also a promising target for therapeutic innovation. The research progress made thus far has laid a strong foundation, but continued interdisciplinary efforts spanning molecular neuroscience, pharmacology, biomaterials, and clinical medicine are essential to fully harness the therapeutic potential of ferroptosis‐targeted strategies in the fight against stroke‐induced brain injury.
Author Contributions
Yilan Fei and Qi Leng: Participated in collecting, assessing, and interpreting the data. Made significant contributions to data interpretation and manuscript preparation. Yilan Fei and Qi Leng: Provided substantial intellectual input during the drafting and revision of the manuscript. All authors have read and approved the final manuscript.
Funding
The authors have nothing to report.
Conflicts of Interest
The authors declare that they have no financial conflicts of interest.
Consent to Publish
The manuscript has neither been previously published nor is under consideration by any other journal. The authors have all approved the content of the paper.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Bi‐Tiao, H. , Y. Yun , Q. R. Lin , X. H. Liu , and Y. D. Liu . 2025. “Targeting Ferroptosis With Natural Products: Investigating Promising Agents for Treating Diabetes and Its Complications.” Biomedicine & Pharmacotherapy 189: 118304.40633206 10.1016/j.biopha.2025.118304 · doi ↗ · pubmed ↗
- 2Briones‐Valdivieso, C. , F. Briones , S. Orellana‐Urzúa , S. Chichiarelli , L. Saso , and R. Rodrigo . 2024. “Novel Multi‐Antioxidant Approach for Ischemic Stroke Therapy Targeting the Role of Oxidative Stress.” Biomedicines 12, no. 3: 501. 10.3390/biomedicines 12030501.38540114 PMC 10968576 · doi ↗ · pubmed ↗
- 3Bu, Z.‐Q. , H.‐Y. Yu , J. Wang , et al. 2021. “Emerging Role of Ferroptosis in the Pathogenesis of Ischemic Stroke: A New Therapeutic Target?” ASN Neuro 13: 17590914211037505. 10.1177/17590914211037505.34463559 PMC 8424725 · doi ↗ · pubmed ↗
- 4Chai, Z. , J. Zheng , and J. Shen . 2024. “Mechanism of Ferroptosis Regulating Ischemic Stroke and Pharmacologically Inhibiting Ferroptosis in Treatment of Ischemic Stroke.” CNS Neuroscience & Therapeutics 30, no. 7: e 14865. 10.1111/cns.14865.39042604 PMC 11265528 · doi ↗ · pubmed ↗
- 5Chen, F. , R. Kang , D. Tang , and J. Liu . 2024. “Ferroptosis: Principles and Significance in Health and Disease.” Journal of Hematology & Oncology 17, no. 1: 41. 10.1186/s 13045-024-01564-3.38844964 PMC 11157757 · doi ↗ · pubmed ↗
- 6Chen, Q. M. , and A. J. Maltagliati . 2018. “Nrf 2 at the Heart of Oxidative Stress and Cardiac Protection.” Physiological Genomics 50, no. 2: 77–97. 10.1152/physiolgenomics.00041.2017.29187515 PMC 5867612 · doi ↗ · pubmed ↗
- 7Chen, T. , L. Ding , M. Zhao , et al. 2024. “Recent Advances in the Potential Effects of Natural Products From Traditional Chinese Medicine Against Respiratory Diseases Targeting Ferroptosis.” Chinese Medicine 19, no. 1: 49. 10.1186/s 13020-024-00918-w.38519984 PMC 10958864 · doi ↗ · pubmed ↗
- 8Deng, L. , S. He , N. Guo , W. Tian , W. Zhang , and L. Luo . 2023. “Molecular Mechanisms of Ferroptosis and Relevance to Inflammation.” Inflammation Research 72, no. 2: 281–299. 10.1007/s 00011-022-01672-1.36536250 PMC 9762665 · doi ↗ · pubmed ↗