Adams-Oliver Syndrome: A Clinical Diagnosis in the Genomic Era
Srinivas GL, Jignesh Sharma, Sabavath Arun, Ajay Vaidya, Amber Kumar

TL;DR
This paper discusses a case of Adams-Oliver Syndrome where genetic testing failed to find a cause, highlighting the importance of clinical diagnosis in rare genetic conditions.
Contribution
The paper emphasizes the ongoing role of clinical evaluation in diagnosing AOS when genomic testing is inconclusive.
Findings
Whole-exome sequencing did not identify any pathogenic variant in a patient with clinical signs of AOS.
The vascular disruption theory is supported as a likely pathogenic mechanism for AOS.
Clinical judgment remains essential for diagnosing AOS and guiding multidisciplinary management.
Abstract
Adams-Oliver syndrome (AOS) is a rare congenital condition marked by defects of the scalp and malformations of the distal extremities. Although several causative genes have been identified, including ARHGAP31, DLL4, NOTCH1, and DOCK6, a subset of clinically suspected cases remains genetically unresolved. We report the case of a four-year-old female patient, the firstborn of a third-degree consanguineous marriage, who presented with a febrile seizure. Physical examination revealed aplasia cutis congenita (ACC) over the parietal scalp, low-set ears, and bilateral lower limb hypoplasia with absent nails, fulfilling two major diagnostic criteria for AOS. Echocardiography revealed a small ostium secundum atrial septal defect, meeting one minor criterion. Whole-exome sequencing (WES) did not identify any pathogenic variant. The child was referred for surgical evaluation of scalp and limb…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
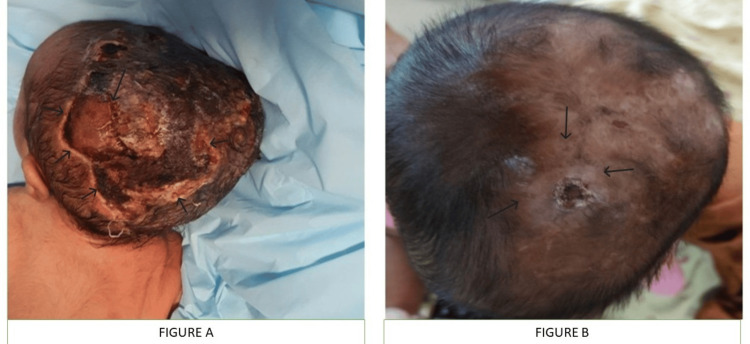 Figure 1
Figure 1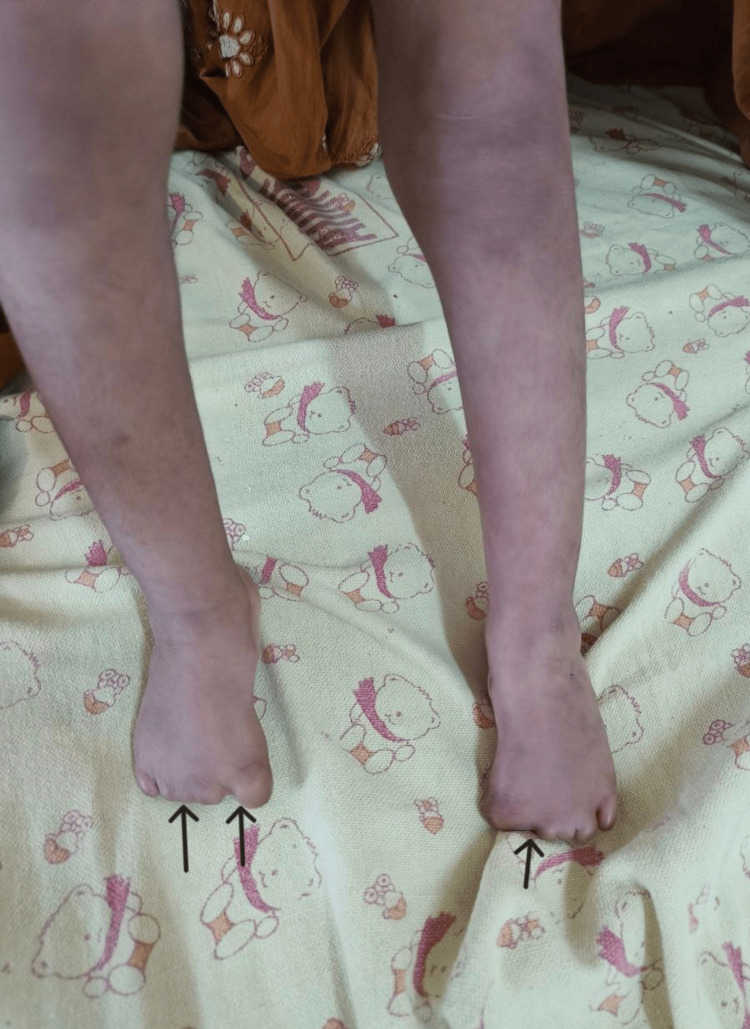 Figure 2
Figure 2| Category | Criteria | Details / Examples |
| Major Criteria | Terminal transverse limb defects | Hypoplastic digits, absent nails, brachydactyly, ectrodactyly, oligodactyly, or more severe distal limb reduction anomalies. |
| Aplasia cutis congenita (ACC) | Most often scalp lesions are noted, may present as solitary or multiple hairless, scarred, or ulcerated areas, sometimes with an underlying bone defect. | |
| Positive family history | Documented diagnosis of Adams-Oliver syndrome (AOS) in first-degree relatives; supports inherited etiology. | |
| Minor Criteria | Cutis marmorata telangiectatica congenita (CMTC) | Persistent, fixed, mottled vascular skin pattern often involving the limbs. |
| Congenital cardiac anomalies | Septal defects and structural malformations such as atrial septal defect (ASD), ventricular septal defect (VSD), or more complex cardiac lesions. | |
| Vascular anomalies | Pulmonary hypertension, intracranial vascular malformations, renal or hepatic vascular defects. | |
| Diagnostic Threshold | Presence of two major criteria, or one major plus one minor criterion | Confirms a clinical diagnosis of AOS. |
| Parameters | Value | Normal range |
| Hemoglobin | 12 | 12-16 g% |
| Total leucocyte count | 12400 | 4000-11000cells/mm3 |
| Platelets | 254000 | 150-450 thousand/microL |
| C-reactive protein | 5.4 | <5 mg/dl |
| Urea | 19 | 20-40 mg/dl |
| Creatinine | 0.4 | 0.6-1.2 mg/dl |
| Sodium | 138 | 135-145 mmol/L |
| Potassium | 3.9 | 3-5 mmol/L |
| Albumin | 3.8 | 3.5-5.5 g/dl |
| Blood culture | Sterile | - |
| Urine culture | Sterile | - |
| Calcium | 8.5 | 8.8-11 mg/dl |
| Phosphate | 4.2 | 2.5-4.5 mg/dl |
| Magnesium | 2.4 | 1.9-2.5 mg/dl |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsSkin and Cellular Biology Research · Dermatological and Skeletal Disorders · Hypertrophic osteoarthropathy and related conditions
Introduction
Adams-Oliver syndrome (AOS) is a striking constellation of congenital anomalies named after the American pediatricians Frederick H. Adams and R. Oliver, who first described it in 1945. It is a rare multisystem disorder primarily characterized by scalp and skull defects, known as aplasia cutis congenita (ACC), alongside terminal transverse limb defects that can range from brachydactyly to complete aplasia of digits or limbs [1]. The eponym reflects the syndrome's initial recognition in a sibling pair with these hallmark cutaneous and skeletal features, establishing it as a distinct entity amid heterogeneous congenital malformation syndromes. The estimated incidence is one in 225,000 live births, though underreporting is likely due to its variable expressivity and overlap with other disorders [2]. Clinically, AOS warrants suspicion in neonates presenting with bitemporal or vertex scalp erosions/ulcers at birth, coupled with symmetric limb reductions, particularly when accompanied by cardiovascular malformations or subtle neurological signs (e.g., microcephaly or seizures), prompting early multidisciplinary evaluation to guide prognosis and management.
While AOS has been linked to autosomal dominant and recessive mutations in multiple genes, including ARHGAP31, DLL4, NOTCH1, RBPJ, DOCK6, and EOGT, which converge on critical developmental pathways such as Notch signaling (essential for vascular and limb patterning) and Rho GTPase regulation (key for cytoskeletal dynamics and tissue morphogenesis), sporadic cases remain well documented [3,4]. Advances in genomic sequencing have expanded our understanding of AOS, but the persistent genetic heterogeneity and negative molecular findings in a significant portion of patients underscore the critical, enduring role of clinical diagnosis, as illustrated by the following case.
Case presentation
A four-year-old female child, the firstborn of a third-degree consanguineous marriage, presented in June 2025 with a generalized tonic-clonic seizure lasting one minute, associated with a high-grade fever. A similar febrile seizure had occurred at 2.5 years of age. Developmental milestones were age-appropriate, and family history was unremarkable.
On examination, the child was hemodynamically stable. Dysmorphic features included a 3 × 4 cm area of ACC over the parietal scalp, presenting as alopecia with underlying scarred tissue and no evidence of skull defect on initial cranial ultrasound (Figure 1); low-set ears; and bilateral lower limb anomalies: the second to fifth toes on both feet were severely hypoplastic, appearing as nubs of approximately 5-7 mm in length, with the great toes shorter than expected (approximately 70% of age-matched norms) but better formed; all toenails were absent bilaterally (Figure 2). Based on the combination of ACC and terminal limb anomalies, the child satisfied two major diagnostic criteria for AOS [4]. Cardiac evaluation revealed a small ostium secundum atrial septal defect (4 mm) with a left-to-right shunt and no evidence of right heart volume overload on echocardiography, thus not requiring immediate intervention and fulfilling a minor diagnostic criterion (Table 1). Laboratory investigations for seizure etiology were normal, and neuroimaging (MRI of the brain) and EEG were unremarkable (Table 2).
Aplasia cutis congenita over the parietal scalp in the neonatal period (A) and healed scalp scar at four years of age (B).
Bilateral terminal limb defects with hypoplastic digits and absent nails involving both lower limbs.
The child was managed conservatively for the acute febrile seizure with antipyretics and hydration, achieving complete resolution of fever and seizure within 48 hours without recurrence during the hospital stay. Given the syndromic presentation and third-degree consanguinity suggesting possible autosomal recessive inheritance, whole-exome sequencing (WES) was performed. WES specifically interrogated known AOS-associated genes (ARHGAP31, DLL4, NOTCH1, RBPJ, DOCK6, EOGT) and identified no pathogenic or likely pathogenic variants, effectively ruling out the most common genetic causes for this presentation. At the three-month follow-up visit in September 2025, the child remained neurologically stable with no further seizures, appropriate weight gain, and stable limb function aided by custom orthotic inserts for gait support. The scalp scar showed complete epithelialization without infection or ulceration. Genetic counseling was provided, and the child was referred to plastic surgery for evaluation of scalp and limb defects, with orthopedic consultation for potential reconstructive options. Multidisciplinary follow-up continues with pediatrics, cardiology (for atrial septal defect (ASD) monitoring), neurology, and genetics every six months to assess for vascular complications and developmental progress.
Discussion
AOS is characterized by a wide phenotypic spectrum ranging from isolated scalp and limb anomalies to multisystem involvement. The major diagnostic criteria include terminal transverse limb defects, ACC, and a positive family history. In contrast, the minor criteria comprise cutis marmorata telangiectatica congenita, congenital heart anomalies, and vascular malformations (Table 1). Diagnosis is supported when two major criteria or one major plus one minor criterion are fulfilled. Our case met the threshold with two major (ACC and limb anomalies) and one minor criterion (ASD) [2,5].
Our patient's phenotype, featuring a 3 × 4 cm parietal ACC without skull defect, severe bilateral lower limb hypoplasia (second and fifth toe nubs, absent nails), and a hemodynamically insignificant ASD, aligns closely with variants commonly associated with NOTCH1 mutations (emphasizing vascular/limb disruptions) or DOCK6 (recessive, multisystem involvement). Yet, no such variants were detected, underscoring AOS's marked genetic heterogeneity and potential for novel loci. Despite substantial advances in next-generation sequencing, a proportion of clinically diagnosed AOS cases remain genetically unresolved, as in our patient. Given the third-degree consanguinity, an autosomal recessive form of AOS is plausible, particularly involving genes such as DOCK6 and EOGT; however, negative WES findings at these and other loci may reflect limitations in detecting deep intronic, regulatory, or structural variants, or suggest involvement of yet-uncharacterized genes [6]. This emphasizes the continued importance of clinical diagnosis, particularly in resource-limited settings. The most widely accepted pathogenic mechanism remains the vascular disruption hypothesis, in which impaired perfusion during embryogenesis results in scalp and limb abnormalities [7]. Additional systemic features, such as cardiac anomalies, CNS malformations, and developmental delay, are variably present [8].
Management of AOS requires a tailored, multidisciplinary approach to address its multisystem features and prevent complications. For ACC diagnosed at birth, initial conservative treatment is preferred for smaller lesions (<5 cm) without exposed bone, involving moist wound care (e.g., hydrogel or foam dressings), infection prophylaxis (e.g., topical antimicrobials like Betadine), and serial monitoring to promote epithelialization and avoid hemorrhage/meningitis risks; surgical options, such as skin grafting or tissue expansion, are reserved for larger defects or those with underlying skull involvement, as in our case where referral for reconstruction was advised post healing [9]. In this patient with bilateral lower limb hypoplasia and incomplete toes, orthopedic management includes custom orthotics/prosthetics for gait support, physical therapy to enhance mobility, and potential reconstructive surgery (e.g., toe lengthening or syndactyly release if webbing develops); early intervention mitigates functional deficits and improves quality of life. Broader care encompasses cardiology follow-up for ASD monitoring, neurology for seizure prophylaxis, and genetic counseling for recurrence risks (up to 50% in dominant forms, variable in recessive forms). Regular surveillance for vascular complications (e.g., pulmonary hypertension) is essential. This case highlights the enduring role of clinical acumen in diagnosing rare syndromes in the genomic era, particularly when advanced testing fails to provide molecular confirmation [10].
Conclusions
We report the case of a child with classical features of AOS fulfilling both major and minor diagnostic criteria. Despite a comprehensive evaluation including WES, no genetic variant was detected. This example highlights the limitations of genomic testing and emphasizes the crucial role of clinical assessment in diagnosing, directing interdisciplinary care, and providing family counselling. Key learning includes the following: AOS should be suspected when ACC occurs with terminal limb defects, with or without systemic involvement; the diagnosis can be made clinically by fulfilling established major and minor criteria. Genetic testing, including WES, may not always identify a pathogenic variant, highlighting the continued importance of detailed phenotyping in the genomic era.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Hereditary deformities in man: due to arrested development J Hered Adams FH Oliver CP 37361945 https://doi.org/10.1093/oxfordjournals.jhered.a 105415
- 2A case of Adams-Oliver syndrome Adv Biomed Res Saeidi M Ehsanipoor F 167620172938767810.4103/2277-9175.221861 PMC 5767801 · doi ↗ · pubmed ↗
- 3Gain-of-function mutations of ARHGAP 31, a Cdc 42/Rac 1 GT Pase regulator, cause syndromic cutis aplasia and limb anomalies Am J Hum Genet Southgate L Machado RD Snape KM 574585882011 https://doi.org/10.1016/j.ajhg.2011.04.0132156529110.1016/j.ajhg.2011.04.013PMC 3146732 · doi ↗ · pubmed ↗
- 4Adams-Oliver syndrome review of the literature: refining the diagnostic phenotype Am J Med Genet A Hassed S Li S Mulvihill J Aston C Palmer S 7908001732017 https://doi.org/10.1002/ajmg.a.378892816041910.1002/ajmg.a.37889 · doi ↗ · pubmed ↗
- 5Adams-Oliver syndrome Int J Dermatol Kutlubay Z PehlivanÖ 352354532014 https://doi.org/10.1111/j.1365-4632.2012.05533.x 2432081810.1111/j.1365-4632.2012.05533.x · doi ↗ · pubmed ↗
- 6Adams-Oliver syndrome: vestigial tail and genetics update Arch Plast Surg Zhu VZ Hansen-Kiss E Hecht JT Payne PE 5175224920223591955610.1055/s-0042-1751107 PMC 9340189 · doi ↗ · pubmed ↗
- 7Vascular pathogenesis of transverse limb reduction defects J Pediatr Hoyme HE Jones KL Van Allen MI Saunders BS Benirschke K 8398431011982 https://doi.org/10.1016/S 0022-3476(82)80343-0713117310.1016/s 0022-3476(82)80343-0 · doi ↗ · pubmed ↗
- 8Adams-Oliver syndrome: a case with full expression Pediatr Rep Dehdashtian A Dehdashtian M 651782016 https://doi.org/10.4081/pr.2016.65172743330710.4081/pr.2016.6517 PMC 4933813 · doi ↗ · pubmed ↗