Confirmation of biallelic VPS11 variants as a cause of complex dystonic syndrome
Arnaud Storck, Marie Thérèse Abiwarde, Gaelle Hardy, Anne-Sophie Lebre, Sophie Scheidecker, Maria Cristina Antal, Mathieu Anheim, Thomas Wirth

TL;DR
This paper confirms that mutations in the VPS11 gene can cause a complex dystonic syndrome, based on a second reported case.
Contribution
The study provides the first replication of adult-onset dystonia linked to biallelic VPS11 variants.
Findings
VPS11 mutations are linked to complex dystonic syndrome in adults.
A second case confirms the association between biallelic VPS11 variants and dystonia.
This expands the known clinical spectrum of VPS11-related disorders.
Abstract
•VPS11 is associated with hypomyelinated leukodystrophy affecting children.•A single case of adult-onset generalized dystonia was reported.•We provide the first replication by describing a second case of complex dystonia related to biallelic variants in VPS11. VPS11 is associated with hypomyelinated leukodystrophy affecting children. A single case of adult-onset generalized dystonia was reported. We provide the first replication by describing a second case of complex dystonia related to biallelic variants in VPS11.
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
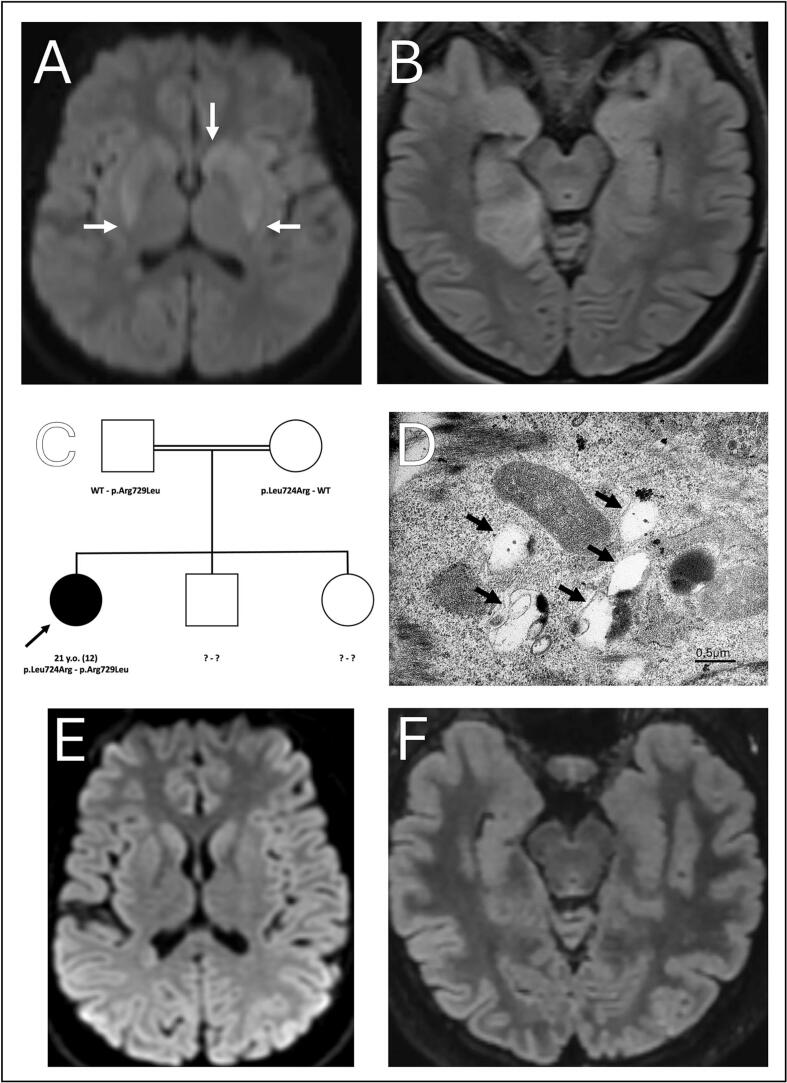 Figure 1
Figure 1Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsRNA regulation and disease · Porphyrin Metabolism and Disorders · Neurological diseases and metabolism
While acknowledged as a cause of hypomyelinated leukodystrophy [1,2], VPS11-associated dystonia was described in a single patient [3].
We report here a case of VPS11-associated dystonia concerning a female patient born at term from a consanguineous union without family history of neurological disorders. Her psychomotor acquisitions were normal until the age of 12 when she developed repeated episodes of paroxysmal gait disturbances lasting few minutes and triggered by exercise. These manifestations halted spontaneously before recurring at the age of 17. Cerebral and medullar MRI were unremarkable. The phenotype subsequently broadened with the occurrence of left-sided myoclonus, pyramidal syndrome of four limbs and cervical dystonia (Video). Levodopa was initiated in the hypothesis of a dopa-responsive dystonia without efficacy.
Her condition worsened following an infection causing the exacerbation of movement disorders. A brain MRI identified FLAIR hypersignal located in the right temporal lobe (Fig. 1). EEG revealed background slowing while a first lumbar puncture showed no anomaly. Aciclovir was started in the hypothesis of a herpetic encephalitis and the patient referred to the Strasbourg University Hospital Neurology department. Further examination revealed pyramidal syndrome of four limbs, left-sided hemidystonia, mild cerebellar ataxia, positive generalized asymmetric asynchronous rest and action myoclonus, and dysphagia (Video). A second lumbar puncture was unremarkable except for tau-protein elevation. Repeated brain MRI revealed anterior putaminal and caudate nucleus head hyperintensity on diffusion-weighted imaging (Fig. 1) showing diminished hyperintensity without full regression at month 30 while a second EEG was consistent with myoclonic status epilepticus (Fig. 1).Fig. 1(A.) Diffusion-weighted hyperintensity in the left caudate nucleus and posterior putamens (arrows). (B.) FLAIR hyperintensity in the right mesial temporal lobe. (C.) Familial pedigree. Current age and age at diagnosis (in parentheses). (D.) Electron microscopy of cultured fibroblasts revealing large mainly clear vacuolar structures (arrows) (E.) June 2023 MRI showing regression of previous diffusion-weighted hyperintensities. (F.) June 2023 MRI showing regression of the previous FLAIR hyperintensity.
Empirical treatments (Supplementary material 1) were administered in the hypothesis of autoimmune encephalitis or inborn error of the metabolism, however unsuccessfully, the patient requiring admission to the Pediatric Intensive Care Unit owing to coma. Detailed description of the therapeutics initiated is available as supplementary. The patient subsequently improved in the following months (Video).
Her clinical milestones can therefore be summarized with an asymptomatic period ranging from 0 to 12 years old briefly followed with the onset of exercice-triggered paroxysmal movement disorder lasting minutes, a subsequent remission from 12 to 17 years old before the onset of cerebellar ataxia, myoclonus, dystonia and pyramidal syndrome with an acute exacerbation leading to coma before gradual recovery. She presented with cerebellar ataxia and spastic paraplegia when last evaluated at 21 years old.
First tier biological tests were unremarkable and a whole genome sequencing was performed in trio with the parents. This research was made possible through access to the data generated by the 2025 French Genomic Medicine Initiative [4]. The patient was found to be compound heterozygous for two missense variants in VPS11. The maternal c.2171 T > G; p.(Leu724Arg) variant was absent from populational database (gnomAD V4.1.0) and predicted pathogenic by various in silico prediction tools while the paternal c.2186G > T; p.(Arg729Leu) variant was still absent from populational database but with conflicting in silico predictions (Fig. 1, supplementary material 2**). Although homozygous variants would have been expected given the consanguinity of the pedigree, indels up to 50 base-pairs were screened using GATK HaplotypeCaller (v4.1.8.0), deletions between 50 base-pairs and 21 kilo-base-airs using Manta (v1.6.0) and copy-number variants larger than 21 kilo-base-pairs using CNVnator (v0.4.1) without the identification of any other structural nor copy-number variants in dystonia-related genes. Electron microscopy of cultured fibroblast biopsy revealed large mainly clear vacuolar structures similar to that of the patient described in Monfrini et al. (Fig. 1)**. Subsequently, the variants were classified as likely pathogenic (class IV ACMG: PS3 + PM2 + PP3) [5].
Our case suggest VPS11 variants may be associated with a complex dystonic syndrome. Notable difference compared to the prior report consists in an unalike disease course with a current complicated hereditary spastic paraplegia phenotype and the absence of basal ganglia T2 hypointensity, which may be related to the age difference between the two cases. Deterioration following an intermittent event (hereby consisting in an undocumented infection) bears resemblance to inborn errors of metabolism. Whether this episode represents an aspect of VPS11-related disorder or a coincidental event will require further evidence.
Given the consistency of clinical, molecular and histological data between these independent cases [3], it appears henceforth acceptable to consider that VPS11-associated clinical spectrum encompasses both, a white-matter pathology and dystonia.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Edvardson S.Gerhard F.Jalas C.Hypomyelination and developmental delay associated with VPS 11 mutation in Ashkenazi-jewish patients J. Med. Genet.5211201574975310.1136/jmedgenet-2015-10323926307567 · doi ↗ · pubmed ↗
- 2Monfrini E.Zech M.Steel D.Kurian M.A.Winkelmann J.Di Fonzo A.HOPS-associated neurological disorders (HOPSAN Ds): linking endolysosomal dysfunction to the pathogenesis of dystonia Brain J. Neurol.144920212610261510.1093/brain/awab 16133871597 · doi ↗ · pubmed ↗
- 3Monfrini E.Cogiamanian F.Salani S.A Novel Homozygous VPS 11 Variant May Cause Generalized Dystonia Ann. Neurol.894202183483910.1002/ana.2602133452836 PMC 8048445 · doi ↗ · pubmed ↗
- 4Abadie C.Abderrahmane A.Abdous O.PFMG 2025–integrating genomic medicine into the national healthcare system in France Lancet Reg Health – Eur.20255010.1016/j.lanepe.2024.101183 PMC 1191079140093400 · doi ↗ · pubmed ↗
- 5Richards S.Aziz N.Bale S.Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology Genet. Med.175201540542310.1038/gim.2015.3025741868 PMC 4544753 · doi ↗ · pubmed ↗