New parajeilongviruses detected in bats but not in humans: assays for screening and diagnostic purposes
Emilia Pulkkinen, Reilly Jackson, Ruut Joensuu, Essi M. Korhonen, Moses Muia Masika, Omu Anzala, Joseph G. Ogola, Paul W. Webala, Tamika J. Lunn, Kristian M. Forbes, Olli Vapalahti, Tuure Kinnunen, Tarja Sironen, Anne J. Jääskeläinen

TL;DR
Researchers developed PCR assays to detect zoonotic paramyxoviruses and found two new parajeilongviruses in bat samples from Kenya.
Contribution
Development of PCR assays for paramyxovirus screening and discovery of new parajeilongviruses in bats.
Findings
Nested-pan-PCR and RT-qPCR assays effectively screened for paramyxoviruses in human and bat samples.
Two new parajeilongviruses were identified in Angolan free-tailed bat samples from Kenya.
No paramyxoviruses were detected in human samples from Finland and Kenya.
Abstract
To enhance preparedness against existing and new zoonotic viruses such as Hendra virus, Nipah virus, and other paramyxoviruses, screening tools and efficient diagnostic methods are needed. Here, we established a conventional nested pan-PCR assay using previously described primers for screening of human and bat samples. Additionally, we developed specific real-time RT-PCR (RT-qPCR) assays to detect Nipah virus and Hendra virus genotypes 1 and 2. Both PCR methods demonstrated good performance and could be used for screening of paramyxoviruses. Human serum and cerebrospinal fluid samples from 558 Finnish patients and 60 serum samples from Kenyan patients were screened using the nested-pan-PCR assay, and all were negative. In addition, we screened 340 synanthropic bat samples collected during 2021 and 2023 from Kenya, resulting in the discovery of two parajeilongviruses in Angolan…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
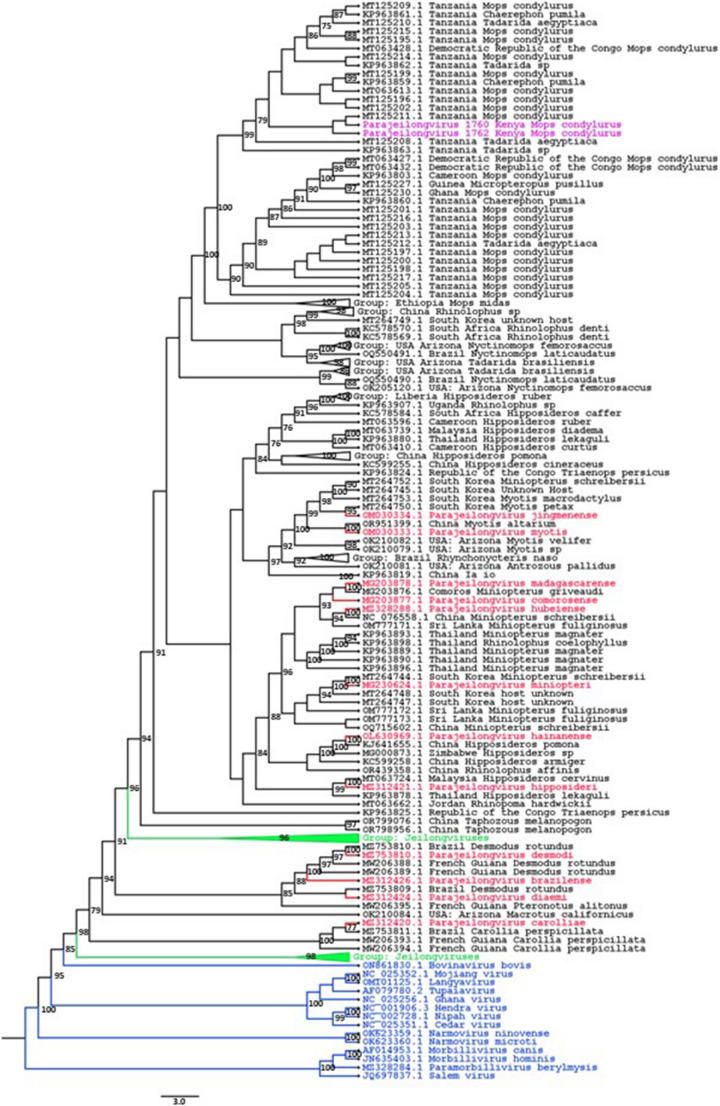 Figure 1
Figure 1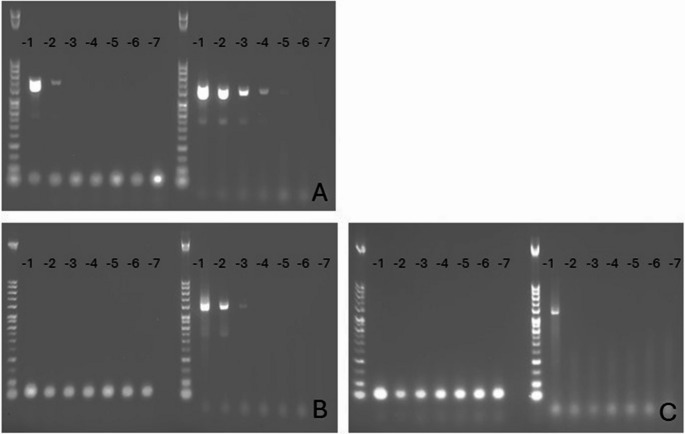 Figure 2
Figure 2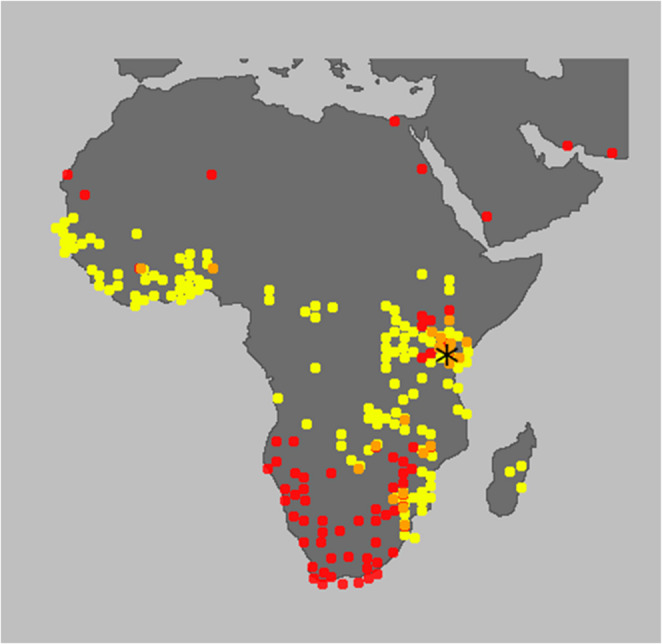 Figure 3
Figure 3- —University of Helsinki (including Helsinki University Central Hospital)
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsVirology and Viral Diseases · Mosquito-borne diseases and control · Respiratory viral infections research
Introduction
Comprehensive preparedness is essential for a rapid response to emerging zoonotic pathogens and future pandemics. Research shows that 60.3% of emerging pathogens have a zoonotic origin, 71.8% of which originate from wildlife [1]. One of the families of high-risk viruses is Paramyxoviridae, in particular, the subfamilies Orthoparamyxovirinae and Feraresvirinae, which both deserve special attention when preparedness is established. These two subfamilies include diverse paramyxoviruses (PMVs) that infect mammals of various species. A notable PMV that infects humans is measles virus (Morbillivirus hominis). This virus spreads extremely efficiently via droplets and still continues to cause childhood mortality worldwide. Measles virus can also cause a wide spectrum of non-fatal complications, including central nervous system (CNS) infections and pneumonia [2]. Less-severe and more-common human PMV infections include respiratory tract infections caused by parainfluenza viruses.
Many PMVs possess zoonotic potential because of their high mutation rate, a typical characteristic of RNA viruses, and their ability to switch host species [3]. Two examples of such zoonotic viruses are the bat-borne viruses Hendra virus (HeV) and Nipah virus (NiV), which have caused severe, often fatal infections in humans in Southeast Asia and Australia [4, 5]. Fruit bats serve as the primary reservoirs for these viruses [6]. However, domesticated animals, such as horses and pigs, have been implicated as intermediate hosts facilitating virus transmission to humans [5]. HeV infections typically manifest as equine-borne sporadic spillover events in Australia, whereas NiV causes annual outbreaks linked to the consumption of contaminated date palm sap or through pigs, for example, in Bangladesh and India [7, 8]. A new emerging paramyxovirus with the pathogenicity of a henipavirus and the transmissibility of measles virus would pose a severe public-health risk, potentially triggering an epidemic – or even a pandemic.
Fruit bats are known to host various paramyxoviruses [9, 10]. For example, in Africa, henipavirus RNAs have been detected in straw-colored fruit bats (Eidolon helvum) in Ghana [11] and the Republic of Congo [12]. Moreover, serological evidence suggests the presence of henipa-like viruses circulating in domesticated pigs from several different pig farms in Uganda [13]. While no henipavirus infections in humans have been reported in Africa, Sosuga virus – a closely related pathogen belonging to the subfamily Rubulavirinae that is in Rousettus aegyptiacus bats in Uganda – has been transmitted to a human, causing acute febrile illness [14]. In Europe, PMVs have been detected in different mammalian species, including insectivorous bats, shrews, and hedgehogs [15–18]. However, spillover events from reservoir animals to humans have not been reported to date. The bat populations in Finland consist only of insectivorous bats [19], and it is currently unknown whether these bat species would carry potentially zoonotic PMVs. To date, no zoonotic PMV infections have been reported in Finland.
In order to study human samples collected from patients in Finland and Kenya with suspected CNS infections or other acute or febrile infections, we established a nested pan-PCR assay using previously designed primers [20] for the detection of PMVs. This assay was also used to screen bat samples from Kenya for the presence of PMVs. As nested assays are prone to contamination, i.e., due to cross-contamination from the positive PCR control, we designed a PCR control with a built-in modification that can be identified by sequencing to avoid false positives in diagnostic settings. For emergency settings, the nested pan-PCR assay is too slow and laborious to be practical. Accordingly, we established fast and specific RT-qPCR methods for the detection and discrimination of NiV and HeV. The HeV RT-qPCR was designed to detect both HeV genotype 1 (G1) and the recently discovered genotype 2 (G2) [21], in order to cover all of the viruses known to cause human disease.
Materials and methods
Human samples
Altogether, 679 samples from Finnish and Kenyan patients were collected and screened. From Finland, 606 serum samples and 13 cerebrospinal fluid samples were collected retrospectively from 558 individuals (median age, 46 years; range, 1–88 years; sex, data not available) between August 2018 and November 2018 from 16 different hospital districts and the Åland Islands. The samples were collected from anonymized sample archives with limited patient data. All samples were from patients with suspected severe acute viral infections, particularly central nervous system infections. Symptoms and differential diagnostics from Finnish patients are listed in Table 1. All data were anonymized, and the research was carried out with ethical approval (permits HUS/32/2018, HUS/211/2020, HUS/244/2021, and 159/HUS/151/2022) from the HUS Diagnostic Center, Helsinki, Finland. A subset of 60 serum samples from 60 Kenyan patients (median age, 19 years; range, 7 months-83 years; sex, 30 female, 29 male, 1 unknown) was obtained from a larger sample collection [22] from Taita-Taveta County, southeastern Kenya, in October 2016. All patients had an acute febrile illness, and the symptoms and differential diagnostics are listed in Table 2. Most of the Kenyan patients had been in contact with either domesticated farm animals or wild animals, including bats and rodents. The characteristics of the participants were reported previously by Masika et al. [22].Table 1. List of differential diagnostics and symptoms of the Finnish patients (N = 558). The list includes tested viral and bacterial agents, and any data available derived from the laboratory data management system. Within approved research permits, only data available in the laboratory data management system could be exploited. No clinical diagnoses of the patients were availableIndividuals with a CSF sample screened for HSV1, HSV2, or VZV nucleic acids****81Positive for HSV21Positive for VZV2Individuals screened for enterovirus nucleic acids****59Positive for enterovirus (other sample types)2Positive for enterovirus from CSF4Individuals screened for borreliosis/neuroborreliosis****131High IgG antibody level, indicating acute/recent borreliosis20Neuroborreliosis (high intrathecal index)16Suspected neuroborreliosis1Individuals screened for TBEV infection****558Acute TBEV infection28Other diagnosis listed in laboratory system****48Encephalitis without a known etiological agent23Acute infection without a known etiological agent16Autoimmune encephalitis1Acute disseminated encephalomyelitis1Demyelinating disease of the central nervous system1Meningitis3Myelitis2Fifth disease1Bacterial infection indicated in the laboratory system CSF3 BSI1 Sinusitis1 Legionellosis1Symptoms of the patientsHeadache, neck pain and/or stiffness19Fever11Visual disturbance6Facial paralysis3Balance disorder2Loss of cognition6Tactile disturbance2Pain4Cramping2Motor weakness2Upper respiratory symptoms1Papilledema1Infertility1SUMMARYAll individuals558No causative agent or diagnosis listed in laboratory system430 (430/558; 77%)Causative agent detected (microbial)80Other diagnosis listed in laboratory system48HSV, herpes simplex virus; VZV, varicella zoster virus; TBEV, tick-borne encephalitis virus; CSF, cerebrospinal fluid; BSI, bloodstream infectionTable 2List of differential diagnostics and symptoms of the Kenyan patients (N = 60)Dengue virus diagnostics****Number of patientsIgG positive, IFA6IgM positive, IFA5IgM positive, ELISA1At least one positive result from dengue virus diagnostics7Chikungunya virus diagnosticsIgG positive, IFA3IgM positive, IFA9IgM positive, ELISA6Sindbis virus diagnosticsIgG positive19IgM positive0Symptoms of the patientsRash1Joint pain28Myalgia24Vomiting10Diarrhea6Headache5Cough9Runny nose9Oral sores2Shivers1Odynophagia1Convulsions1Jaundice1Dysuria2Pharyngitis1Nausea1IFA, immunofluorescence assay; ELISA, enzyme-linked immunosorbent assayAll assays are described in more detail by Masika et al. [19]
RNA was extracted from human samples using a semi-automated MagNA Pure 96 System (Roche) and a Total Nucleic Acid Kit according to the manufacturer’s instructions. Extracted RNA samples were stored at −70°C.
Bat samples
All bat samples were collected in Taita-Taveta County in southeastern Kenya between August 2021 and May 2023. Permission to conduct bat fieldwork was granted through permits from the National Commission for Science, Technology and Innovation (#NACOSTI/P/21/9267), the Kenya Wildlife Service (#KWS/BRM/500 and WRTI/RP/118.6), and the University of Arkansas Institutional Animal Care and Use Committee (#22012).
Bats were captured in flyways, building roosts, and inside roosting buildings, using mist-nets or by hand. Bats were placed into individual cotton bags upon capture and stored in a humid cool location overnight. The following morning, the species, sex, and age of the bats were recorded (Table 3). Fecal samples were collected from inside the bags and directly from the excreting bats during the sampling process. This capturing protocol followed previously described standards [23]. At dusk on the day after capture, the bats were returned to the collection site and released. The fecal pellets were stored in 2-ml tubes containing 0.5 ml of RNAlater, first at −20°C for a short initial period, and then at −80°C for long-term storage. Later, a subset of 340 samples was selected for RNA extraction using Invitrogen TRIzol Reagent (Thermo Fisher Scientific, Waltham, Massachusetts, USA) according to the manufacturer’s guidelines.Table 3. Screened bat species, the number of samples screened (total N = 340) and the number of positive samples, age, and sex distribution of the individual batsBat speciesn/positivesLittle free-tailed bat (Mops pumilus)136/0Angolan free-tailed bat (Mops condylurus)132/2Heart-nosed bat (Cardioderma cor)31/0Andrew Rebori's house bat (Scotophilus andrewreborii)13/0Banana serotine (Afronycteris nanus)6/0Hinde's lesser house bat (Scotoecus hindei)5/0Egyptian fruit bat (Rousettus aegyptiacus)4/0Wahlberg's epauletted fruit bat (Epomophorus wahlbergii)3/0Sundevall’s roundleaf bat (Hipposideros caffer)3/0Yellow-winged bat (Lavia frons)2/0Silvered bat (Glauconycteris argentata)1/0Geoffroy's horseshoe bat (Rhinolophus clivosus)1/0Lander’s horseshoe bat (Rhinolophus landeri)1/0Egyptian free-tailed bat (Tadarida aegyptiaca)1/0African trident bat (Triaenops afer)1/0Age and sex distribution of individual bats****Age Adult343/2 Juvenile24/0Sex Female208/0 Male177/2
Nested pan-PCR assay and screening
Previously designed PCR primers [20] were used to establish a nested pan-PCR protocol for detection of different members of the family Paramyxoviridae. In addition, for this assay, we designed a PCR control with an artificial built-in modification as a contamination control to avoid false positives (see Appendix). According to the taxonomy nomenclature at the time of publication, the viruses amplified with these PCR primers included members of the genera Henipavirus, Morbillivirus, Respirovirus, and Rubulavirus. Under the current taxonomy, the viruses detected using these primers would be classified in several genera of the two subfamilies Orthoparamyxovirinae and Feraresvirinae. Using these primers and various PMV controls, PCR products ranging in length from 600 to 700 base pairs were produced and then sequenced by the Sanger method. As the protocol was established for diagnostic purposes, it was further tested with human serum and CSF samples. To enhance the specificity of the assay for clinical samples, the level of nonspecific amplification of human DNA was reduced by using a touch-down PCR as the second PCR cycle, by first allowing flexible primer binding followed by more-specific amplification conditions towards the end of the procedure (see Appendix).
To test the performance of the nested pan-PCR assay, viral RNA from measles virus, canine morbillivirus (previously canine distemper virus, CDV), HeV G1, and NiV were used. Because HeV and NiV are classified as biosafety-level-4 pathogens, they were received as inactivated virus stocks from the Commonwealth Scientific and Industrial Research Organisation (CSIRO, Australia). In addition, we designed a cDNA plasmid as an assay contamination control. This plasmid contained part of the genome of CDV, with a recognizable insertion of artificial and non-coding nucleotides (CDV plasmid; Integrated DNA Technologies) to further discriminate possible contamination from the original viruses (Supplementary Fig. S1). To assess the sensitivity of the nested pan-PCR assay, a tenfold dilution series of CDV plasmid was tested, as well as RNAs from HeV G1 and NiV (Fig. 2).
All human and bat samples were screened using the nested pan-PCR assay, followed by agarose gel electrophoresis (AGE), sequencing and annotation using the core nucleotide collection from the National Centre for Biotechnology Information (NCBI) with Basic Local Alignment Tool (BLAST).
Henipavirus RT-qPCR assays
Specific RT-qPCR assays for NiV and HeV G1 and G2 were established using the CFX96 platform (Bio-Rad) (see Appendix). Primers were designed to recognize the region of the genome encoding the nucleocapsid protein. All of the RT-qPCR assays were validated using viral RNA of NiV and HeV G1, commercial quantified plasmid controls of HeV G1 (IDT) and G2 (IDT), and a negative control panel (Table 4). The sensitivity was determined by calculating the limit of detection (LOD) and intra-assay repeatability for 10 parallel reactions.Table 4. Cross-testing of henipavirus RT-qPCR assaysHendra-RT-qPCR (result: neg/pos)Nipah-RT-qPCR (result: neg/pos)Virus RNACanine distemper virus (in-house)NegNegMeasles virus (vaccine strain)NegNegYellow fever virus (17D, vaccine strain)NegNegYellow fever virus (Asibi, vaccine strain)NegNegZika virus (strain MR766)NegNegDengue virus 1 (Zeptometrix)NegNegDengue virus 3 (Zeptometrix)NegNegReston ebolavirus (PHE)**NegNegTai Forest virus (PHE)**NegNegMarburg virus (Musoke, PHE)**NegNegBundibugyo virus (PHE)NegNegZaire ebolavirus (PHE)NegNegLassa virus (RKI)NegNegLymphocytic choriomeningitis virus (in-house)NegNegCrimean-Congo hemorrhagic fever virus (RKI)NegNegRift-Valley fever virus (RKI)NegNegParainfluenza 4 virus (in-house)NegNegRinovirus 1B (in-house)NegNegMetapneumovirus (in-house)NegNegRespiratory syncytial virus (in-house)NegNegHendra virus (CSIRO)PosNegNipah virus (CSIRO)NegPosPlasmidsCommercial plasmid-based construct: Nipah (IDT); 4.4E + 8 copies per reactionNegPosCommercial plasmid-based construct: HeV G1 (IDT); 4.3E + 8 copies per reactionPosNegCommercial plasmid-based construct: HeV G2 (IDT); 8.4E + 8 copies per reaction*PosNegRespiratory samples, altogether N = 9Rhinovirus positive, N = 6Neg (6/6)Neg (6/6)Rhino-, adeno- and parainfluenza virus 4 positive, N = 1Neg (1/1)Neg (1/1)Rhino- and enterovirus positive, N = 1Neg (1/1)Neg (1/1)Enterovirus positive, N = 1Neg (1/1)Neg (1/1)Other sample types, negative*Serum, N = 20Neg (20/20)Neg (20/20)CSF, N = 5Neg (5/5)Neg (5/5)EDTA-blood, N = 20Neg (20/20)Neg (20/20)*Challenging the RT-qPCR with high load of control; Cycle values of 13.9 and 10.6 for commercial Nipah and Hendra plasmid controls, respectively. IDT, Integrated DNA Technologies; CSF, cerebrospinal fluid; CSIRO, Commonwealth Scientific and Industrial Research Organisation (Australia). **Viral RNAs kindly provided by the Public Health Institute (PHE, Porton Down, UK) and the Robert Koch Institute (RKI, Germany). *** Blood samples were originally from previously healthy individuals screened for human herpesvirus 6 nucleic acids.
Results
Nested pan-PCR testing of human samples
In total, 606 serum and 13 CSF samples from Finnish patients, as well as 60 serum samples from Kenyan patients (total N = 679) were screened using the nested pan-PCR assay. PCR products (~ 650 base pair long) were obtained from 16 sera (16/666, 2.4%) and two CSF samples (2/13, 15.4%), all of which were successfully sequenced. No viral nucleic acids were detected among the sequenced amplicons, and since all of these sequences corresponded to human DNA, they were interpreted as nonspecific background amplifications. No PMVs were detected in the Finnish and Kenyan patient samples.
Nested pan-PCR testing of bat samples
In total, 340 bat fecal samples from 340 individuals comprising 16 different bat species (Table 3) from Taita-Taveta County (Kenya) were screened by nested pan-PCR, and amplification products of PMVs were detected in two fecal samples of Angolan free-tailed bats (Mops condylurus). The positive fecal samples were collected from two adult male bats in May 2023 from the main building of an orphanage house. The 457-base-pair-long sequences were first annotated using the BLAST annotation tool, followed by construction of a Bayesian maximum-likelihood phylogenetic tree using IQ-TREE [24, 25], which was visualized using FigTree [26] (Fig. 1 and Supplementary Fig. S2). This analysis revealed that these sequences shared a high degree of similarity with viral sequences from the genera Jeilongvirus and Parajeilongvirus of the subfamily Orthoparamyxovirinae. The highest sequence identity values – 99.11% and 98.66% – were observed with partial parajeilongvirus sequences obtained from M. condylurus samples from Tanzania. In addition, these sequences shared a high degree of similarity with partial viral sequences from free-tailed bats (Tadarida sp.). Unfortunately, no remaining sample material was available for further NGS sequencing attempts with these two positive bats.Fig. 1. Maximum-likelihood phylogenetic tree based on partial sequences of L gene of the two detected parajeilongviruses and related viruses with midpoint rooting. The tree was constructed using sequences from orthoparamyxoviruses (blue), jeilongviruses (green), and parajeilongviruses (red), as well as the 100 top BLAST hits (black) for the two detected parajeilongviruses (purple). Strains found in the official ICTV taxonomy are labeled with their taxonomic nomenclature, whereas the sequences identified using BLAST are named using their geographical location and host species. The best-fitting model was chosen for these 475-base-pair-long sequences using IQTREE ModelFinder [35]. The analysis was performed in IQTREE [36], using the GTR + F + R6 model with 1000 bootstrap replicates, and the tree was visualized using FigTree v1.4.4 [26]. A full non-collapsed tree is shown in Supplementary Fig. S2)
Extracted RNA from CDV, measles virus, HeV, NiV, and a commercial plasmid control for CDV with a built-in contamination control were all successfully amplified using the nested pan-PCR and were subsequently sequenced and correctly annotated (see Appendix). The negative control and negative panel were all negative; no amplification was detected by AGE using these CSF or blood samples. Tenfold dilution series of CDV plasmid and HeV and NiV RNAs showed increased sensitivity with the nested pan-PCR assay compared to the initial PCR step of the assay (Fig. 2). The CDV control demonstrated clear amplification already after the first PCR step, whereas HeV and NiV could be detected only after the nested step. The nested PCR step increased performance by 1000-fold with the CDV plasmid control (Fig. 2).Fig. 2. Dilution series of CDV plasmid and Nipah virus and Hendra virus RNAs using agarose gel electrophoresis (AGE). The performance between the first round of PCR followed by the nested round of PCR was compared using a tenfold dilution series of CDV plasmid control (IDT; panel A), as well as Nipah virus RNA (CSIRO; panel B) and Hendra virus RNA (CSIRO; panel C). The PCR products with a size of ~ 650 bp from the first and second PCR cycle are shown on the left and right, respectively. All PCR runs also included negative and CDV-positive controls (not shown in these AGE images). CSIRO, Commonwealth Scientific and Industrial Research Organisation (Australia)
Henipavirus RT-qPCR assays
The assays were validated using extracted viral RNA from inactivated HeV and NiV (CSIRO) and commercial quantified plasmid controls of NiV, HeV G1, and HeV G2 (IDT). The henipavirus RT-qPCR assays were able to discriminate NiV from HeV. A negative panel comprised of 14 sera, five cerebrospinal fluid samples, 20 EDTA-blood samples, and nine respiratory samples, in addition to a panel of 20 non-henipaviral RNAs (Table 4), were all negative (68/68, 100%). By using 95% confidence intervals (SPSS, Probit, IBM), the LOD for the henipavirus RT-qPCRs was 14.1, 7.2, and 9.8 copies per PCR reaction for HeV G1 (IDT), HeV G2 (IDT), and NiV (IDT), respectively. At a copy level of 430–440 with 10 parallel reactions for HeV G1, the average Ct value was 33.4, the standard deviation (STDEV) was 1.1, and the coefficient of variation (CV) was 3%. At a copy level of 8400 for HeV G2, the Ct value was 35.5, the STDEV was 0.78, and the CV was 2%, and for NiV at a copy level of 430–440, the Ct value was 32.5, the STDEV was 0.70, and the CV was 2%.
Discussion
In Finland, no endemic zoonotic infections caused by PMVs have been reported to date. However, another PMV – measles virus – is still causing sporadic infections almost annually. The highest rate of measles virus infections was in 2011, with 27 laboratory-confirmed cases [27]. The coverage of measles, mumps, and rubella (MMR) vaccination varies geographically in Finland but is high overall [27]. In this study, as expected, we did not find measles virus or other PMVs in the Finnish samples (N = 619). The Kenyan patient samples were collected from April to August 2016, before the large-scale measles-rubella vaccination campaign started in October 2016 in Kenya. Nevertheless, we did not find any measles virus or other PMVs in the Kenyan samples. However, it is worth noting that we did not have any vaccination data from these 60 Kenyan patients. Although measles vaccines have been used for decades in Kenya, measles virus continues to cause infections every year. For example, in 2016, the vaccination coverage for the first and second dose of the measles vaccine was 93% and 35%, respectively. In the same year, 128 measles cases were reported in Kenya[28]. Of note, there is currently a measles epidemic ongoing in the USA [29].
Complete genome sequences have been obtained for paramyxoviruses from bat samples collected in Kenya, including samples from Triaenops, Hipposideros, Coleura, Miniopterus, Otomops, Nycteris, Rousettus, Cardioderma and Chaerephon sp., but none have been obtained from Mops species [30, 31]. In our study, a parajeilongvirus was detected in two individual Mops condylurus fecal samples, using nested pan-PCR and sequencing. The sequences from these bat samples exhibited high sequence similarity (90–99% identity) to other partial genome sequences of putative paramyxoviruses found in different bat species, i.e., Mops condylurus, Mops pumilus, and Tadarida sp., in northern Africa (Fig. 1). Partial genome sequences of parajeilongviruses found in Tanzania, obtained from the NCBI GenBank database, were 98–99% identical to our sequences, suggesting the circulation of this parajeilongvirus type in the African Great Lakes region. The similarity of the viral sequences in Mops sp. and closely related Tadarida sp. suggests potential for spillover between these species. However, due to the lack of whole genome sequences, and because only a highly conserved part of the sequence was used to study the relationships among different virus strains, no definitive conclusions are possible. Although Tadarida aegyptiaca is generally found in more-southern parts of Africa than M. condylurus, they are both present in the African Great Lakes region, especially in Kenya (Fig. 3) [32, 33].Fig. 3. Map highlighting the geographical distribution of Mops condylurus (yellow) and Tadarida aegyptia (red) and their region of overlap (orange). The sample collection site is indicated by an asterisk [32, 33]
Taxonomically, the genus Parajeilongvirus was separated from the genus Jeilongvirus in 2023 based on differences in their host associations and genomic structure (International Committee on Taxonomy of Viruses). In the current taxonomy, all of the parajeilongviruses originate mostly from bats, which is consistent with our findings, since the sequences obtained from M. condylurus samples were more similar to parajeilongviruses than jeilongviruses (Fig. 1). Both genera consist of newly discovered viruses, and therefore, in-depth research data on these viruses is limited. Although new viruses are detected continually, the most common screening methods in use result in short nucleotide sequences from conserved areas of the genome, leaving many open questions, including the detailed characteristics of the detected viruses.
Overall, the procedure for the nested pan-PCR assay fits well into a routine diagnostic setting, and in this study, no cross-contamination was detected. The nested PCR step includes a contamination control that appears to be suitable for routine diagnostic purposes. The assay produced some background amplification (human DNA), more frequently with CSF samples (15.4%) than with serum samples (2.4%) , but this was technically straightforward to exclude by Sanger sequencing. The process of implementing a plasmid control improved the practicality of the laborious contamination-prone nested PCR in routine diagnostics, highlighting its potential application, for example, in resource-limited settings where RT-qPCR or next-generation sequencing may not be available. More nonspecific amplification was detected by AGE when using samples from bats than when using human samples. This was expected, as bat fecal samples typically include a greater diversity of background nucleic acids than human blood or CSF samples.
For emergency settings, molecular methods based on nested PCR and sequencing are not recommended due to their time-consuming nature and susceptibility to cross-contamination. For situations in which results are needed within hours rather than days, RT-qPCR assays with a short turnaround time and easily interpreted results should be designed. In this study, we designed, performed, and validated specific RT-qPCR assays for NiV, HeV G1, and HeV G2. These assays demonstrated good performance, including efficient detection of the new HeV G2. These methods are now available for use in clinical diagnostics. Although RT-qPCR assays are efficient, conventional PCR assays combined with Sanger sequencing remain valuable tools for distinguishing new virus variants and genotypes from existing ones [21].
Preparedness for zoonotic infections requires basic research and surveillance to enhance our understanding of viral circulation in humans and wildlife. The family Paramyxoviridae is likely to include numerous undiscovered zoonotic pathogens. A recent example of a spillover transmission from animals to humans is the shrew-borne Langya virus, which infected 35 people in China in 2022 [34]. Due to global travel and constant human interactions, pandemic risk assessment must extend beyond national borders. Rapid and efficient clinical tools, such as qPCR, for diagnosing travel-associated infections, combined with the surveillance of local zoonotic risks in wildlife, are essential for preventing emerging virus infections and viral transmission. The paramyxovirus surveillance methods described here were used for screening both human and bat samples and could be coupled with full-genome sequencing when possible. In the future, these methods can be applied to other wildlife samples, including those from rodents and shrews, to further uncover the diversity of paramyxoviruses and to assess potential local zoonotic risks in Finland, Kenya, and elsewhere.
Electronic Supplementary Material
Below is the link to the electronic supplementary material
Supplementary File 1 (DOCX 778 KB)
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1World Health Organization (WHO) https://www.who.int/health-topics/measles#tab=tab_1.
- 2Finnish Institute for Health and Welfare Finnish National Infectious Diseases Register. https://thl.fi/aiheet/infektiotaudit-ja-rokotukset/seurantajarjestelmat-ja-rekisterit/tartuntatautirekisteri/tartuntatautien-esiintyvyystilastot/tartuntatautien-esiintyvyys-suomessa-raportit