Deciphering comprehensive profiles of pathogenies and resistome of pork using integrating metagenomic and isolation strategies
Lianwei Ye, Qiao Hu, Tao Zang, Yaling Wang, Heng Heng, Edward Wai Chi Chan, Sheng Chen

TL;DR
This study explores the pork microbiome to identify harmful pathogens and antibiotic resistance genes using a combination of isolation and sequencing methods.
Contribution
The paper introduces an integrated approach combining isolation and metagenomic sequencing to analyze pork microbiomes comprehensively.
Findings
A high diversity of pathogens and resistance genes was found on pork surfaces.
Potential sources of contamination include air, transportation, water, and cross-contamination.
The study highlights the need for multifaceted food surveillance strategies.
Abstract
The pork microbiome was investigated using an integrated approach combining isolation and metagenomic sequencing methods to comprehensively analyze the pathogens and resistome on pork surfaces. The study revealed a large number and diversity of pathogens and resistance genes, potentially originating from air, transportation, water, or cross‐contamination. These findings underscore the importance of implementing multifaceted food surveillance strategies to monitor and mitigate these risks effectively.
Click any figure to enlarge with its caption.
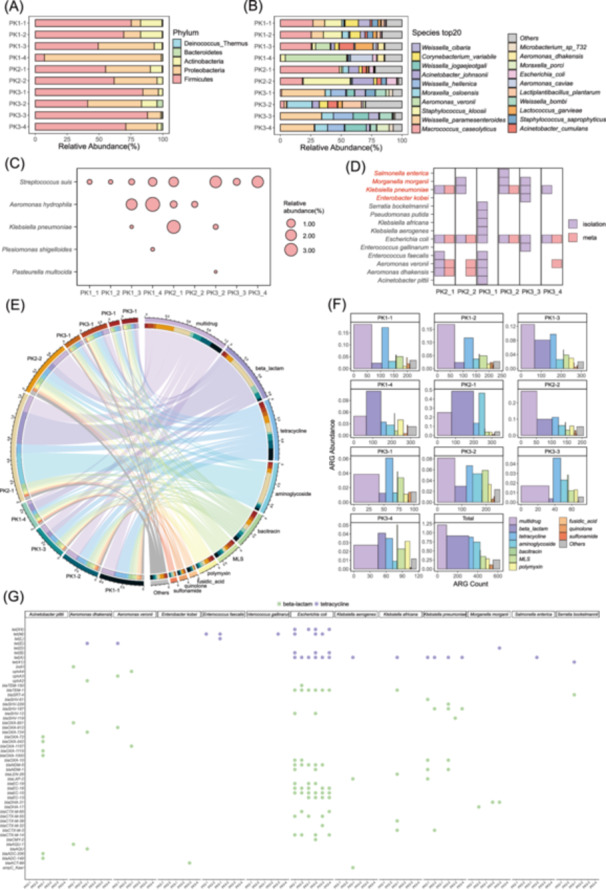 Figure 1
Figure 1 Figure 2
Figure 2- —The General Research Fund of Research Grant Council of the Government of Hong Kong SAR
- —The Theme‐based Research Scheme
- —The Key Research and Development Program of Guangdong Province
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsMicrobial infections and disease research · Bacteriophages and microbial interactions · Aquaculture disease management and microbiota
To the editor,
Pork is a significant source of foodborne pathogens, contributing to food poisoning. Meat products, including chicken, pork, and beef, are among the most common sources of pathogens, with global consumption projected to reach 376 million tons by 2030 [1, 2, 3]. Pork, compared to other meats, carries a higher risk of antimicrobial‐resistant bacteria, particularly Klebsiella pneumoniae and Enterococcus spp., which are resistant to critical antibiotics. While poultry is linked to Campylobacter and Salmonella, pork is associated with multidrug‐resistant Enterococcus and Klebsiella, increasing the risk of foodborne infections. This highlights pork's unique food safety risks compared to beef or poultry. Some pathogens carry antimicrobial resistance genes (ARGs), posing a multidrug resistance (MDR) threat. For instance, Salmonella strains isolated from pork in Jiangsu [4], China, exhibited high resistance to aminoglycosides (88.21%), tetracyclines (90.24%), and florfenicol (91.87%), harboring genes such as strA, bla _ TEM‐1 _, and floR. Traditional isolation methods have been instrumental in identifying human pathogenic bacteria (HPB) on pork, including Escherichia coli, Salmonella enterica, Vibrio parahaemolyticus, and Staphylococcus aureus [5, 6, 7, 8, 9]. These approaches have revealed ARGs like bla _ CMYs _, mcr‐1, and novel plasmids, underscoring their role in uncovering new ARG variants [9, 10, 11, 12]. However, metagenomic sequencing offers a more comprehensive approach to studying microbial diversity. While 16S metagenomic studies, such as those by Koo et al. [13], identified abundant bacteria like Pseudomonas in ground pork, limited research exists on pork surface microbiomes using next‐generation sequencing. Challenges, such as extracting bacterial DNA from pork surfaces, hinder broader application. In our study, we integrated isolation and metagenomic methods to analyze the microbiome and resistome on the surface of pork from Hong Kong wet markets. Among 10 samples, Firmicutes and Proteobacteria were dominant, with 5 HPBs identified. Metagenomic analysis revealed 13 metagenome‐assembled genomes (MAGs), including pathogens like Staphylococcus kloosii. Isolation‐based methods identified 77 strains spanning 14 species, with E. coli and Klebsiella spp. predominating. Notably, some species like Enterococcus spp. and S. enterica were absent in metagenomic data but identified through isolation. Novel species, such as Enterobacter kobei and Klebsiella africana, were reported on pork surfaces for the first time. Metagenomic analysis uncovered 23 ARG types (621 subtypes), including clinically significant ones like bla _ NDMs _ and tet(X4). Isolation methods revealed 16 ARG types (134 subtypes), with 81 unique to this approach. These findings highlight the co‐occurrence of ARGs and virulence factor genes (VFGs) in HPBs, underscoring the complexity of antimicrobial resistance in foodborne pathogens.
RESULTS AND DISCUSSION
1
Microbial communities of pork surface microbiome
Metagenomic analysis identified the microbial communities on pork surfaces collected from wet markets in Hong Kong. Ten samples collected from MongKok Market, Ho Hing Sun Kee Meat Company, and New Generation Fruits & Vegetables were analyzed (Table S1), revealing Firmicutes and Proteobacteria as the dominant phyla, although their prevalence varied across the samples (Figure 1A). Firmicutes dominated most samples, except for PK1‐4 and PK3‐2, where Proteobacteria prevailed. Actinobacteria exceeded 10% abundance in PK1‐1, PK2‐1, and PK3‐2 (Table S2). At the genus level (Table S3), Weissella, Staphylococcus, and Moraxella were consistently dominant. Genera such as Microbacterium, Acinetobacter, Aeromonas, Escherichia, Macrococcus, and Corynebacterium were detected in more than eight samples. The number of genera per sample ranged from eight (PK1‐4) to 45 (PK3‐3). A high diversity of species is observed, with each sample carrying different species, which vary in their relative abundances (Figure 1B). HPB was detected in all samples, comprising five species (Figure 1C). Pathogens such as K. pneumoniae, Enterococcus faecalis, and others appeared sporadically.
Comprehensive analysis of microbial diversity and antibiotic resistance in pork samples. (A) Phylum: A stacked bar chart representing the relative abundance of phylum found in different samples. (B) Species TOP20: A similar stacked bar chart as in (a), but detailing the top 20 species. (C) Human pathogenic bacteria (HPB) Species relative abundance (%): A bubble chart indicating the percentage of HPB species' relative abundance in various samples. (D) A heatmap showing the presence or absence of pathogens in different samples based on the isolation method (Purple indicates the species detected through isolation, while pink indicates the species identified via metagenomic analysis). HPB species are shown in red. (E) Antimicrobial resistance genes (ARGs) Relationships: A circular plot representing the relationships between different ARG types and sample locations, with lines connecting them indicating their associations. The outer ring of the circle is divided into segments labeled with different ARG types and sample locations. (F) ARG Abundance: A bar chart showing the abundance of ARGs and the number of types of ARGs subtypes in each sample. (G) Distribution of ARGs in Isolates: Colored markers within each column represent different ARG genes identified in bacterial strains from that sample. Strains include Acinetobacter pittii、Aeromonas dhakensis、Aeromonas veronii、Enterobacter kobei、Enterococcus faecalis、Enterococcus gallinarum、Escherichia coli、Klebsiella aerogenes、Klebsiella africana、Klebsiella pneumoniae、Morganella morganii、Pseudomonas putida、Salmonella enterica and Shewanella bockelmannii.
Isolation methods identified 76 strains of four pathogenic bacteria across six samples. Dominant species included Klebsiella spp., with isolates such as E. kobei, K. africana, and Salmonella enterica (Figure 1D). Sample PK3‐1 hosted the highest diversity with nine strains, while PK2‐1 and PK3‐3 had five strains each. Isolated strains like E. faecalis and E. gallinarum were absent in metagenomic data, indicating that the isolation method could detect low‐abundance bacteria. While E. coli was detected in all samples by isolation, it was noted in metagenomic data only in some. Similarly, K. pneumoniae showed discrepancies between the two methods. Our combined analysis of pork surfaces revealed K. pneumoniae and other human pathogenic bacteria, which aligns with the findings of Emamjomeh et al. [14], who detected Klebsiella at the genus level. However, our species‐level identification uncovered greater diversity, revealing additional pathogens not detected by traditional methods. The discrepancy between isolation and metagenomic detection of strains like Enterococcus spp. and K. aerogenes could be due to their low abundance in metagenomic datasets. While metagenomics offers a broad overview, isolation detects low‐abundance or fastidious species [15]. Combining both methods provides a more comprehensive understanding of microbial diversity and food safety risks.
Examination of abundance and diversity of ARGs
Metagenomic analysis of pork microbiomes revealed a high diversity and abundance of ARGs, identifying 23 primary ARG types and 621 subtypes (Table S4). Among these, multidrug, beta‐lactam, and tetracycline resistance genes were the most prevalent, with beta‐lactam resistance genes, particularly bla _ NDM _, bla _ OXA _, and bla _ CTX‐M _ subtypes, dominating across samples (Figure 1E,F). Colistin resistance genes (mcr‐1 to mcr‐5) were also detected, albeit at lower relative abundances. Diversity analysis showed significant variation in ARG profiles between samples, reflecting heterogeneity in microbial communities influenced by environmental factors and meat processing conditions. To complement the metagenomic findings, 76 bacterial isolates from pork samples underwent genomic characterization, revealing additional resistance determinants (Figure 1G). Notably, plasmid‐borne genes, including bla _ NDM‐5 _, bla _ CTX‐M‐15 _, mcr‐1, and tet(X4), were detected, signifying their potential for horizontal gene transfer (Figure 2A–P). Bla _ NDM‐5 _, a carbapenemase gene, confers resistance to carbapenems, underscoring its significance in foodborne pathogens. Tet(X4), associated with tigecycline resistance, raises concerns about its emergence in foodborne and clinical contexts.
Distribution, location, and phylogenetic analysis of antimicrobial resistance genes (ARGs) in Isolation and MAGs. (A–P) ARG distribution by antibiotic class: This panel series illustrates the distribution and location of various ARGs associated with different classes of antibiotics. Each panel (A–P) represents a specific class of antibiotics. The y‐axis lists the ARGs, while colored symbols indicate the location and methods used to identify these genes. A diamond symbol represents ARGs located in plasmids. Color coding: purple indicates genes detected only from isolated strains, while red indicates genes detected in both isolated strains and metagenome‐assembled genomes (MAGs).
Furthermore, the comparative analysis demonstrated distinct ARG compositions between metagenomic datasets and isolated genomes, highlighting the value of combining both approaches for comprehensive ARG profiling. These findings underscore the pork microbiome as a reservoir of ARGs, some of which are clinically significant and associated with MDR. The identification of plasmid‐borne bla _ NDM _ and mcr genes, in particular, raises concerns about the potential dissemination of resistance traits through the food chain. This finding highlights the need for routine surveillance to prevent ARG dissemination through foodborne pathogens, posing risks to public health and emphasizing the importance of stringent antimicrobial stewardship in livestock farming. Additionally, some ARGs in the bacteria detected in our study may not expressed, which could be due to environmental factors or the specific conditions under which these bacteria were isolated. While these unexpressed ARGs may pose a lower immediate risk, they could potentially be activated in different environments, such as the human gut, contributing to antimicrobial resistance. Further investigation into the expression of these genes is needed to assess their full impact on public health.
Examination of abundance and diversity of VFGs and MGEs in foodborne pathogens on pork surfaces
Metagenomic analysis identified 13 types of VFGs across all samples, encompassing functions such as adherence, biofilm formation, immune modulation, and stress survival mechanisms, with subtypes ranging from 208 to 1017 (Tables S5, S6). PK1‐1, PK1‐2, and PK2‐1 exhibited over 606 VFG genes, whereas PK3‐1, PK3‐3, and PK3‐4 had significantly fewer. Six types of mobile genetic elements (MGEs), including integrative and conjugative elements, transposons, and insertion sequences, were detected, with PK1‐2, PK1‐3, and PK2‐1 containing over 1000 subtypes, while PK3‐3 had only 411. Dominant MGE genes included ISAs31, Tn6216, and ISAba8. Isolate‐based analysis revealed diverse VFG and MGE profiles across bacterial species, with E. coli showing the highest diversity (10 VFG types, 3841 subtypes). A total of 200 VFG genes, primarily from the effector delivery system, were uniquely identified in isolates. Species such as K. aerogenes and S. enterica harbored distinct genes like allA, hilC, and pltB. Unique MGEs, including Tn3411 in Klebsiella spp. and Tn558 in E. faecalis plasmids, were observed. Comparatively, isolate genomes revealed 55 MGE subtypes not found in metagenomic data, emphasizing their complementarity. The diversity of MGEs and VFGs highlights their role in bacterial adaptation and pathogenicity. Identifying unique genes, such as hilC in S. enterica, underscores the potential for horizontal gene transfer and persistence in foodborne bacteria. Future research will investigate the roles of VFGs and MGEs in bacterial adaptation and virulence, with an emphasis on their potential for horizontal gene transfer and persistence in foodborne pathogens.
MAGs on the surface of pork
Thirteen MAGs were recovered from five samples: PK1‐1 (n = 5), PK1‐2 (n = 4), PK1‐3 (n = 1), PK1‐4 (n = 2), and PK3‐4 (n = 1) (Figure S1). Most MAGs exhibited over 70% completeness and less than 10% contamination, spanning three phyla and six genera: Corynebacterium, Lactiplantibacillus, Weissella, Macrococcus, Staphylococcus, and Escherichia. Two MAGs of Corynebacterium variabile were recovered from PK1‐1 and PK1‐2, carrying the icl VFG. Lactiplantibacillus plantarum, assembled from PK3‐4, is a common foodborne pathogen, while Weissella cibaria, detected in PK1‐1, is typically associated with food fermentation. Three MAGs of Macrococcus caseolyticus, identified from different samples at the same location, included one carrying the tet(M) antibiotic resistance gene. Similarly, three MAGs of Staphylococcus kloosii were identified, with one harboring the blaZ gene. Lastly, a single E. coli MAG from PK1‐4 contained 49 VFGs and exhibited notable genomic diversity. These MAGs reveal diverse microbial populations on pork surfaces, including pathogens and commensals. The 13 MAGs presented in this study, including Corynebacterium variabile and Weissella cibaria, may play a role in foodborne contamination. While these species are not commonly associated with foodborne illness, the presence of VFGs within their genomes, such as icl, suggests that they could contribute to pathogenicity in the food chain. In our future studies, we will focus on characterizing unclassified MAGs and their genomes to explore their potential roles in antimicrobial resistance, virulence, and overall contributions to the microbiome on pork surfaces.
Finally, wet markets are monitored through periodic inspections of hygiene, food handling, and pathogen testing. However, routine surveillance of ARGs, VFGs, and resistant pathogens is lacking. We recommend implementing molecular screening for ARGs and VFGs, enforcing stricter hygiene regulations, and enhancing consumer education to mitigate risks associated with wet market food.
AUTHOR CONTRIBUTIONS
Lianwei Ye: Conceptualization; methodology; software; data curation; investigation; validation; writing—original draft; formal analysis; visualization. Qiao Hu: Investigation; validation. Tao Zang: Investigation. Yaling Wang: Investigation. Heng Heng: Data curation; software. Edward Wai Chi Chan: Writing—review and editing.
CONFLICT OF INTEREST STATEMENT
The authors declare no conflicts of interest.
ETHICS STATEMENT
No animals or humans were involved in this study.
Supporting information
Figure S1. Phylogenetic tree of Isolation and MAGs.
Table S1. Collection sites and geographical coordinates of pork samples. Table S2. Relative abundance of various bacterial phyla across different samples. Table S3. Relative abundance of various bacterial genus across different pork samples (%). Table S4. Antibiotic resistance profile of pork samples. Table S5. Mobile element gene profile of pork samples. Table S6. Virulence gene profile of pork samples.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Heredia, Norma , and Santos García . 2018. “Animals as Sources of Food‐Borne Pathogens: A Review.” Animal Nutrition 4: 250–255. 10.1016/j.aninu.2018.04.006 30175252 PMC 6116329 · doi ↗ · pubmed ↗
- 2Morales‐Partera, Ángela M , Fernando Cardoso‐Toset , Francisco Jurado‐Martos , Rafael J. Astorga , Belén Huerta , Inmaculada Luque , Carmen Tarradas , and Jaime Gómez‐Laguna . 2017. “Survival of Selected Foodborne Pathogens on Dry Cured Pork Loins.” International Journal of Food Microbiology 258: 68–72. 10.1016/j.ijfoodmicro.2017.07.016 28759797 · doi ↗ · pubmed ↗
- 3Abay, Seçil , Reyhan Irkin , Fuat Aydin , Hamit Kaan Müştak , and Kadir Serdar Diker . 2017. “The Prevalence of Major Foodborne Pathogens in Ready‐To‐Eat Chicken Meat Samples Sold in Retail Markets in Turkey and the Molecular Characterization of the Recovered Isolates.” LWT‐Food Science and Technology 81: 202–209. 10.1016/j.lwt.2017.03.052 · doi ↗
- 4Li, Yan , Kangkang Li , Kai Peng , Zhiqiang Wang , Hongqin Song , and Ruichao Li . 2022. “Distribution, Antimicrobial Resistance and Genomic Characterization of Salmonella Along the Pork Production Chain in Jiangsu, China.” LWT 163: 113516. 10.1016/j.lwt.2022.113516 · doi ↗
- 5Xu, Yating , Zhiwei Zheng , Lianwei Ye , Edward Wai‐Chi Chan , and Sheng Chen . 2023. “Identification and Genetic Characterization of Conjugative Plasmids Encoding Coresistance to Ciprofloxacin and Cephalosporin in Foodborne Vibrio spp.” Microbiology Spectrum 11: e 01032–01023. 10.1128/spectrum.01032-23 37395663 PMC 10434038 · doi ↗ · pubmed ↗
- 6Liu, Xiaobo , Ruichao Li , Zhiwei Zheng , Kaichao Chen , Miaomiao Xie , Edward Wai‐Chi Chan , Shu Geng , and Sheng Chen . 2017. “Molecular Characterization of Escherichia coli Isolates Carrying mcr‐1, fos A 3, and Extended‐Spectrum‐β‐Lactamase Genes from Food Samples in China.” Antimicrobial Agents and Chemotherapy 61: e 00064‐17. 10.1128/aac.00064-17 28373188 PMC 5444122 · doi ↗ · pubmed ↗
- 7Liu, Xiaobo , Shu Geng , Edward Wai‐Chi Chan , and Sheng Chen . 2019. “Increased Prevalence of Escherichia coli Strains from Food Carrying bla NDM and mcr‐1‐bearing Plasmids That Structurally Resemble Those of Clinical Strains, China, 2015 to 2017.” Eurosurveillance 24: 1800113. 10.2807/1560-7917.ES.2019.24.13.1800113 30940314 PMC 6446510 · doi ↗ · pubmed ↗
- 8Li, Yinghui , Qiang Luo , Xiaolu Shi , Yiman Lin , Yaqun Qiu , Dongyue Lv , Yixiang Jiang , et al. 2017. “Phenotypic and Genotypic Characterization of Clinical Enterotoxigenic Escherichia coli Isolates from Shenzhen, China.” Foodborne Pathogens And Disease 14: 333–340. 10.1089/fpd.2016.2233 28537439 · doi ↗ · pubmed ↗