Hyperosmolar Hyperglycemic State With Reversible Encephalopathy in a Young Female on Chronic Steroids and Opioids: A Case Report
Athena Myrou

TL;DR
A young woman on long-term steroids and opioids developed a rare hyperglycemic condition with brain symptoms, highlighting the risk in atypical populations.
Contribution
This case report presents HHS with reversible encephalopathy in a young adult with chronic steroid and opioid use, a rare clinical scenario.
Findings
The patient exhibited severe hyperglycemia and encephalopathy despite being young and having no typical diabetes history.
Neuroimaging ruled out acute intracranial pathology, and the patient fully recovered with fluid and insulin therapy.
Chronic steroid use suppressed the HPA axis, and opioid exposure may have contributed to the metabolic crisis.
Abstract
Hyperosmolar hyperglycemic state (HHS) is uncommon in young adults and may signal complex metabolic and pharmacologic interactions, particularly in the context of chronic glucocorticoid and opioid exposure. We describe a 22-year-old female with morbid obesity, idiopathic intracranial hypertension treated with ventriculoperitoneal shunting and neuromodulation, chronic high-dose steroid use, and opioid exposure who presented with severe hyperglycemia, dehydration, headache, and transient confusion. Laboratory findings revealed glucose level of 583 mg/dL, calculated osmolality of approximately 318 mOsm/kg, preserved C-peptide indicating severe insulin resistance, and suppressed adrenocorticotropic hormone and cortisol consistent with exogenous steroid-induced hypothalamic-pituitary-adrenal axis suppression. Urine ketones were present, but serum ketones were absent, supporting HHS without…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
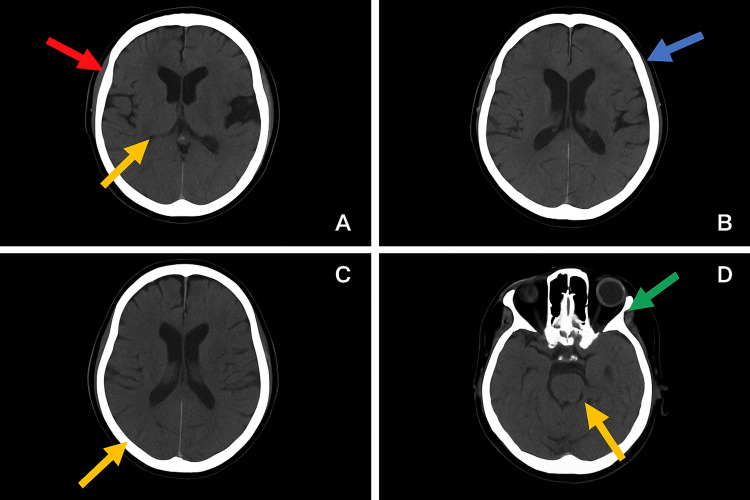 Figure 1
Figure 1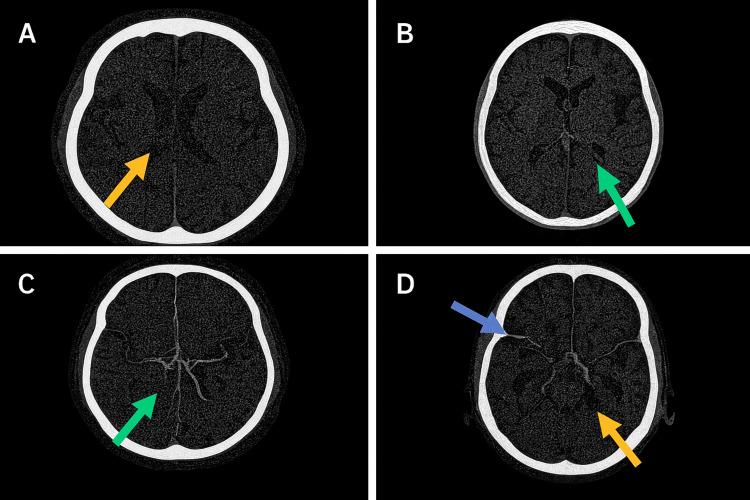 Figure 2
Figure 2| Parameter | Value | Reference range | Interpretation |
| Glucose | 583 mg/dL | 74–100 | Severe hyperglycemia |
| Sodium (measured) | 135 mmol/L | 136–146 | Pseudohyponatremia |
| Sodium (corrected)* | 142.7 mmol/L | — | True sodium level in HHS |
| Potassium | 4.6 mmol/L | 3.5–5.1 | Normal |
| Urea | 70 mg/dL | 17–43 | Dehydration/pre-renal |
| Creatinine | 0.82 mg/dL | 0.66–1.9 | Normal |
| Glycosylated hemoglobin (HbA1c) | 9.9% | 4.0–6.0 | Chronic dysglycemia |
| C-peptide | 5.77 ng/mL | 1.1–4.4 | Severe insulin resistance |
| Urine ketones | ++/+++ | Negative | No DKA |
| Serum osmolality (calculated)** | ~318 mOsm/kg | 275–295 | Hyperosmolar state |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsNeurological and metabolic disorders · Autoimmune Neurological Disorders and Treatments · Neurological Complications and Syndromes
Introduction
Hyperosmolar hyperglycemic state (HHS) is a life-threatening metabolic emergency characterized by severe hyperglycemia, hyperosmolality, dehydration, and altered mental status, typically affecting older adults with type 2 diabetes [1,2]. Its occurrence in young adults is unusual and may indicate underlying endocrine, metabolic, or pharmacologic triggers.
Chronic glucocorticoid exposure is a recognized cause of secondary diabetes and HHS due to enhanced hepatic gluconeogenesis, peripheral insulin resistance, and impaired β-cell function [3,4]. Prolonged supratherapeutic steroid use may also suppress the hypothalamic-pituitary-adrenal (HPA) axis, increasing susceptibility to metabolic instability and infection [5-7]. Opioid exposure may further obscure diagnosis by altering mental status and masking neurologic symptoms [8,9]. Idiopathic intracranial hypertension (IIH) is strongly associated with obesity [10], and intracranial neuromodulation hardware may complicate neurologic assessment and neuroimaging interpretation.
In this patient, the coexistence of IIH further complicated the clinical picture, as IIH-related symptoms such as headache and visual changes may overlap with metabolic or neurologic manifestations of HHS.
This case underscores the importance of maintaining a high index of suspicion for HHS in young adults with significant metabolic and iatrogenic risk factors and highlights the need for coordinated multidisciplinary management.
Case presentation
A 22-year-old female with morbid obesity (BMI = 56.6 kg/m²), IIH treated with a ventriculoperitoneal shunt and occipital nerve stimulator (device model/manufacturer not documented in available records), and chronic supratherapeutic glucocorticoid exposure for presumed inflammatory disease presented to the emergency department with progressive headache, polydipsia, severe hyperglycemia, and transient confusion. Her history also included chronic opioid use with prior tramadol dependence. Her analgesic regimen consisted of long-term use of multiple opioid and centrally acting agents, including tramadol up to 450 mg/day, tapentadol (Palexia) 75 mg every 10 hours, and additional muscle relaxants (Miorel, Viloxidon) and Maroxim, taken regularly since 2021.
On arrival, she was hypertensive (180/120 mmHg) and tachycardic (113 bpm) with normal oxygen saturation and was alert and oriented without focal neurological deficits (Glasgow Coma Scale score = 15/15). Physical examination revealed cushingoid features, including centripetal obesity, violaceous striae, and edema.
Initial laboratory evaluation demonstrated serum glucose at 583 mg/dL, measured sodium at 135 mmol/L (corrected 142.7 mmol/L), urea at 70 mg/dL, creatinine at 0.82 mg/dL, and glycosylated hemoglobin (HbA1c) of 9.9%. Urine ketones were ++/+++ with absent serum ketonemia, and C-peptide was elevated (5.77 ng/mL), suggesting preserved endogenous insulin secretion and severe insulin resistance. Serum osmolality was calculated at ~318 mOsm/kg. Adrenocorticotropic hormone (ACTH) and morning cortisol were suppressed, consistent with exogenous steroid-induced HPA-axis suppression. Mild transaminitis and neutrophilic leukocytosis were noted with low inflammatory markers, likely steroid-attenuated (Table 1).
Neuroimaging with non-contrast CT and CT angiography/venography revealed no acute intracranial pathology or venous thrombosis. Imaging quality was partly limited by the neuromodulation device. No clinical or radiologic evidence of shunt malfunction was identified (Figures 1, 2).
Non-contrast CT of the brain.Axial non-contrast CT images of the brain (A–D) demonstrating no intracranial hemorrhage, no abnormal parenchymal densities, and no midline shift. The ventricular system and subarachnoid spaces appear within normal limits. Mild widening of the subarachnoid space at the vertex is noted. A partially empty sella is present. Several ethmoidal air cells show opacification, while the remaining paranasal sinuses are well aerated. Impacted maxillary teeth projecting into the maxillary sinus floor are visualized. Linear hyperdense subcutaneous structures in the occipital region create streak artifacts.A: Red arrow = widening of the subarachnoid space at the vertex; yellow arrow = partially empty sella.B: Blue arrow = prominent cortical sulci.C: Yellow arrow = prominence of subarachnoid space.D: Green arrow = opacification of ethmoidal air cells; yellow arrow = impacted maxillary tooth projecting into the maxillary sinus floor.
Axial CT angiography images of the brain.A: Normal opacification of the anterior circulation, with symmetric filling of the distal internal carotid arteries and proximal middle cerebral arteries (yellow arrow). No arterial stenosis or occlusion is present.B: The middle cerebral artery branches demonstrate normal contrast enhancement without evidence of vascular narrowing or filling defects (green arrow).C: Continued visualization of patent middle cerebral artery branches with preserved caliber and branching pattern (green arrow).D: Normal opacification of the posterior circulation, including the basilar artery and posterior cerebral arteries (yellow arrow). The superficial cortical veins over the convexity also appear normally opacified (blue arrow). No aneurysm, arteriovenous malformation, or venous thrombosis is identified.
Management and clinical course
The patient was diagnosed with HHS without ketoacidosis and was treated with aggressive isotonic fluids, intravenous insulin, electrolyte management, and continuation of glucocorticoids with a cautious taper to avoid adrenal crisis. During hospitalization, she experienced a brief episode of transient encephalopathy that resolved spontaneously without new findings on repeat imaging. A transient bacteremia was identified and treated according to culture sensitivities.
She received high-dose intravenous hydrocortisone in multiple daily doses over a 13-day period, followed by gradual tapering according to clinical response.
Her neurologic and metabolic status progressively improved. She was transitioned to basal insulin (glargine) with correctional rapid-acting insulin and discharged with close endocrine and neurology follow-up. Written informed consent was obtained from the patient for publication of this case.
Discussion
This case illustrates the interplay of metabolic, endocrine, neurologic, and pharmacologic factors leading to HHS in a young adult. Morbid obesity, prolonged supratherapeutic glucocorticoid exposure, and chronic opioid use produced a high-risk environment for metabolic decompensation [11,12]. The markedly elevated C-peptide level indicated profound insulin resistance rather than insulin deficiency, consistent with steroid- and obesity-driven mechanisms [3,4].
Glucocorticoids are frequently implicated in hyperglycemic crises due to their effects on gluconeogenesis, lipolysis, and peripheral insulin sensitivity [3,4]. Long-term exposure additionally suppresses the HPA axis, blunts inflammatory responses, and predisposes patients to infection and metabolic deterioration [5-7]. Obesity further amplifies systemic inflammation, worsens insulin resistance, and increases vulnerability to hyperglycemic emergencies [13].
Neurologic manifestations of HHS are mediated by hyperosmolality, dehydration, and electrolyte imbalance [2,10]. In this case, opioid exposure is likely to have contributed to fluctuating mental status and confounded neurologic assessment [14,15]. Despite ketonuria, the absence of serum ketonemia, preserved acid-base status, and normal bicarbonate levels made diabetic ketoacidosis (DKA) unlikely [2,16]. In contrast, the patient’s markedly elevated effective osmolality (~318 mOsm/kg) met the diagnostic threshold for HHS as defined by current American Diabetes Association (ADA) and Joint British Diabetes Societies for Inpatient Care (JBDS) guidelines [2,10]. These parameters, together with preserved endogenous insulin secretion, strongly favored the diagnosis of HHS over DKA. The presence of bacteremia in the setting of low inflammatory markers exemplifies the immunosuppressive effects of chronic corticosteroid therapy [17].
Opioid exposure may have contributed to the patient’s transient encephalopathy through several mechanisms. Activation of μ-opioid receptors produces central nervous system (CNS) depression, blunts arousal pathways, and can impair attention and vigilance even at therapeutic doses [18]. In the context of hyperosmolarity, neuronal dehydration and osmotic shifts may further reduce cortical excitability, potentially amplifying opioid-related suppression of consciousness [19]. The combination of μ-receptor-mediated CNS depression and hyperosmolar metabolic stress, therefore, provides a plausible explanation for the patient’s transient alteration in mental status.
Management aligned with HHS guidelines, emphasizing careful fluid resuscitation, intravenous insulin administration, electrolyte monitoring, and continuation of glucocorticoids with a controlled taper to avoid adrenal crisis [2,10]. The reversible encephalopathy observed here highlights the importance of early recognition and prompt metabolic correction.
This report has several limitations inherent to single-patient case studies, which limit the generalizability of the observations. Medication adherence, including the timing and dosage of opioid intake, could not be fully verified because documentation relied partially on patient self-report. Consequently, the temporal relationship between opioid exposure and the brief episode of encephalopathy remains uncertain. Additionally, incomplete historical records regarding the neuromodulation device and long-term steroid use constrain the precision of mechanistic interpretation. Despite these limitations, the case provides valuable clinical insights into the complex metabolic and neurologic interactions in young adults with significant iatrogenic and pharmacologic risk factors.
Conclusions
The present case highlights an uncommon occurrence of HHS in a young adult without known diabetes, precipitated by severe insulin resistance from morbid obesity and prolonged supratherapeutic glucocorticoid exposure, with additional contribution from opioid-associated central nervous system vulnerability. Early recognition of the hyperosmolar state, despite the presence of ketonuria, was essential in differentiating HHS from DKA and guiding appropriate therapy. The coexistence of steroid-induced metabolic dysregulation, HPA-axis suppression, and blunted inflammatory responses created a permissive environment for metabolic crisis and transient encephalopathy. Opioid exposure further complicated neurologic assessment and may have contributed to fluctuating mental status during hospitalization. Prompt fluid resuscitation, insulin therapy, infection management, and a carefully controlled steroid taper facilitated full neurological and metabolic recovery. This case underscores the importance of maintaining a high index of suspicion for HHS in young adults with overlapping endocrine, pharmacologic, and iatrogenic risk factors, and highlights the critical role of multidisciplinary management in optimizing outcomes.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Management of diabetes and hyperglycaemia in the hospital Lancet Diabetes Endocrinol Pasquel FJ Lansang MC Dhatariya K Umpierrez GE 174188920213351549310.1016/S 2213-8587(20)30381-8PMC 10423081 · doi ↗ · pubmed ↗
- 2Hyperglycemic crises in adults with diabetes: a consensus report Diabetes Care Umpierrez GE Davis GM El Sayed NA 125712754720243905290110.2337/dci 24-0032 PMC 11272983 · doi ↗ · pubmed ↗
- 3Steroid diabetes: from mechanism to treatment?Neth J Med van Raalte DH Diamant M 6272722014 https://pubmed.ncbi.nlm.nih.gov/24659588/24659588 · pubmed ↗
- 4Diagnosis and management of adrenal insufficiency Lancet Diabetes Endocrinol Bancos I Hahner S Tomlinson J Arlt W 216226320152509871210.1016/S 2213-8587(14)70142-1 · doi ↗ · pubmed ↗
- 5European Society of Endocrinology and Endocrine Society joint clinical guideline: diagnosis and therapy of glucocorticoid-induced adrenal insufficiency J Clin Endocrinol Metab Beuschlein F Else T Bancos I 1657168310920243872404310.1210/clinem/dgae 250PMC 11180513 · doi ↗ · pubmed ↗
- 6Long-term usage of oral glucocorticoids leading to adrenal insufficiency: a comprehensive review of the literature Cureus Monge Chacón AG Wang C Waqar D Syeda SA Kumar R Meghana DR 015202310.7759/cureus.38948 PMC 1025796937309331 · doi ↗ · pubmed ↗
- 7The global burden of metabolic disease: data from 2000 to 2019 Cell Metab Chew NWS Ng CH Tan DJH 4144283520233688928110.1016/j.cmet.2023.02.003 · doi ↗ · pubmed ↗
- 8Trends in insulin resistance: insights into mechanisms and therapeutic strategy Signal Transduct Target Ther Li M Chi X Wang Y Setrerrahmane S Xie W Xu H 216720223579410910.1038/s 41392-022-01073-0PMC 9259665 · doi ↗ · pubmed ↗