Selective blockade of microRNA-31-5p/calcitonin receptor interaction reverses established atrial fibrosis and atrial arrhythmia substrate
Jasha Trompf, Kathryn Cox, Mohit Hulsurkar, Lucia M Moreira, Chi Him Kendrick Yiu, Aaron M Johnston, Satadru K Lahiri, Shuai Zhao, Paul Robinson, Roddy Hiram, Mozhdeh Mehdizadeh, Lorena Perez Carrillo, Rana Sayeed, George Krasopoulos, Vivek Srivastava, Nicholas Walcot

TL;DR
This study shows that blocking a specific microRNA interaction can reverse heart tissue scarring and arrhythmia linked to atrial fibrillation.
Contribution
The study introduces a novel RNA-based therapy that reverses established atrial fibrosis and arrhythmia substrate.
Findings
AF patients show upregulation of miR-31–5p in atrial cardiofibroblasts.
Blocking miR-31–5p/CTR interaction reverses advanced atrial fibrosis and arrhythmogenesis in vivo.
The therapy selectively increases CTR expression and targets fibrogenesis.
Abstract
Atrial fibrillation (AF), the commonest cardiac arrhythmia, is a major contributor to mortality and morbidity. Atrial tissue fibrosis, a hallmark of structural remodelling in AF, is currently incurable and significantly hinders AF-treatment. MicroRNA(miR)-31 is linked to ageing (a key risk factor for AF). Here, we show that AF-patients are characterised by upregulation of miR-31–5p in atrial cardiofibroblasts that negatively regulates the calcitonin receptor (CTR), thereby promoting atrial fibrogenesis and arrhythmia. Specific blockade of miR-31–5p/CTR-mRNA binding with LNA-miRNA-Target-Site-Blocker selectively increases atrial CTR expression and reverses advanced atrial fibrosis and arrhythmogenesis in vivo. These findings suggest a key role for miR-31–5p/CTR binding in promoting atrial fibrosis and arrhythmogenesis, and represents a first example of an RNA-based therapeutic capable of…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
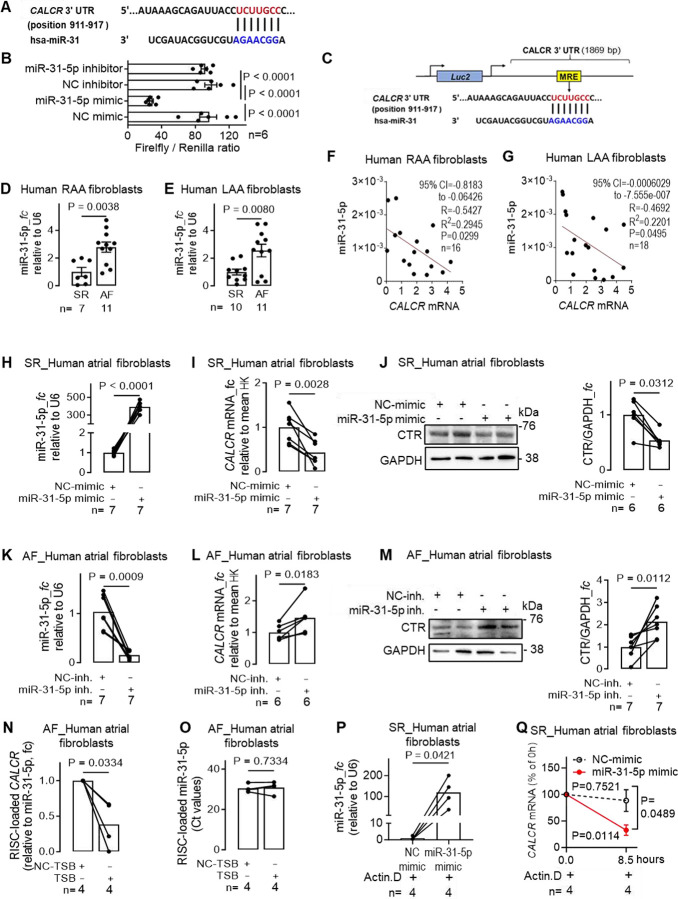 Figure 1
Figure 1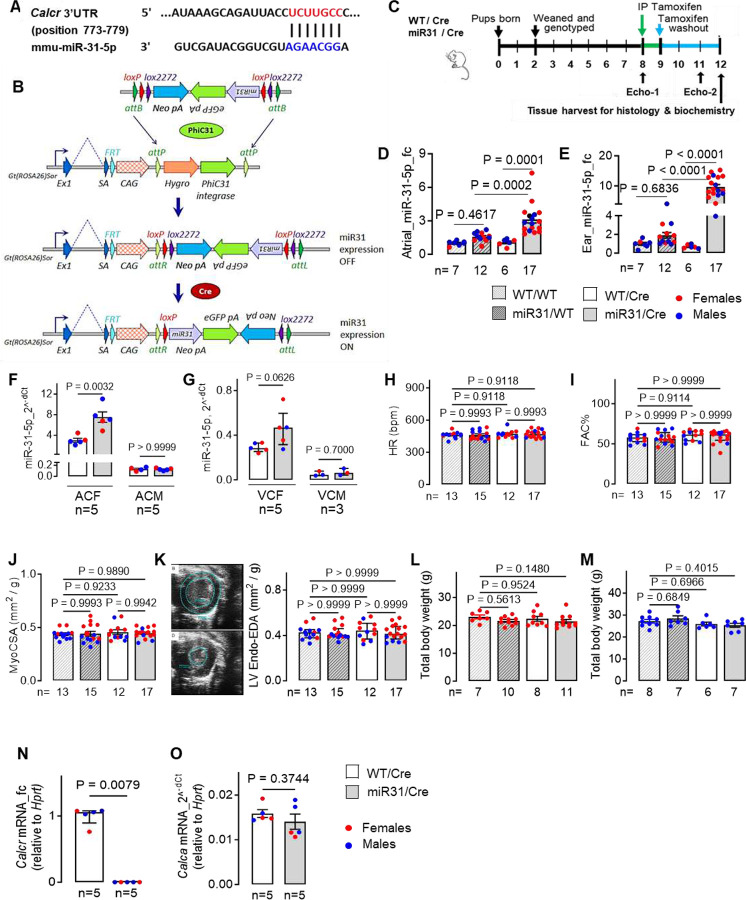 Figure 2
Figure 2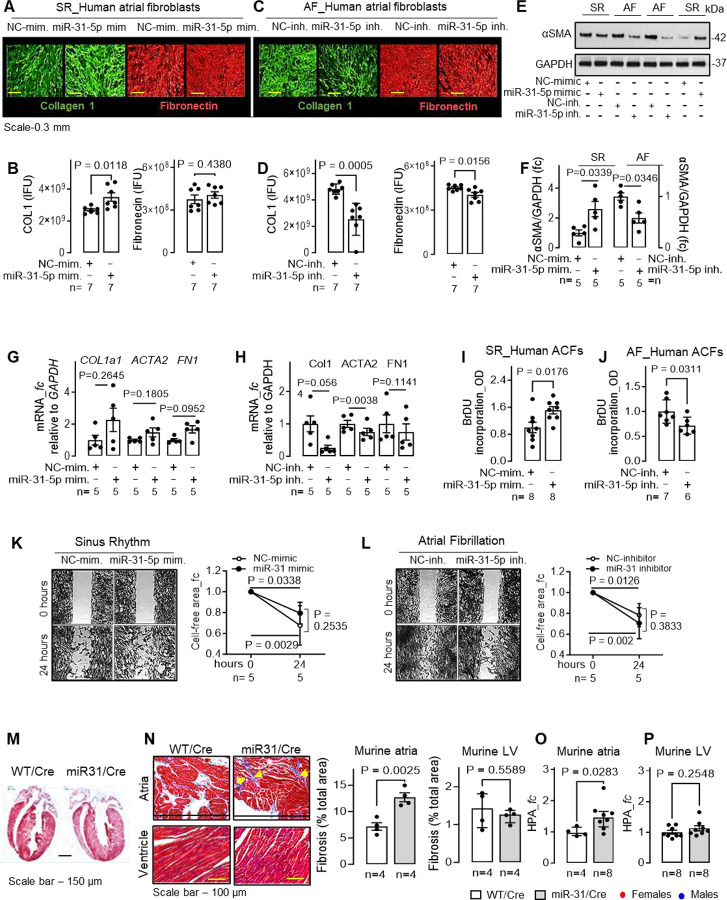 Figure 3
Figure 3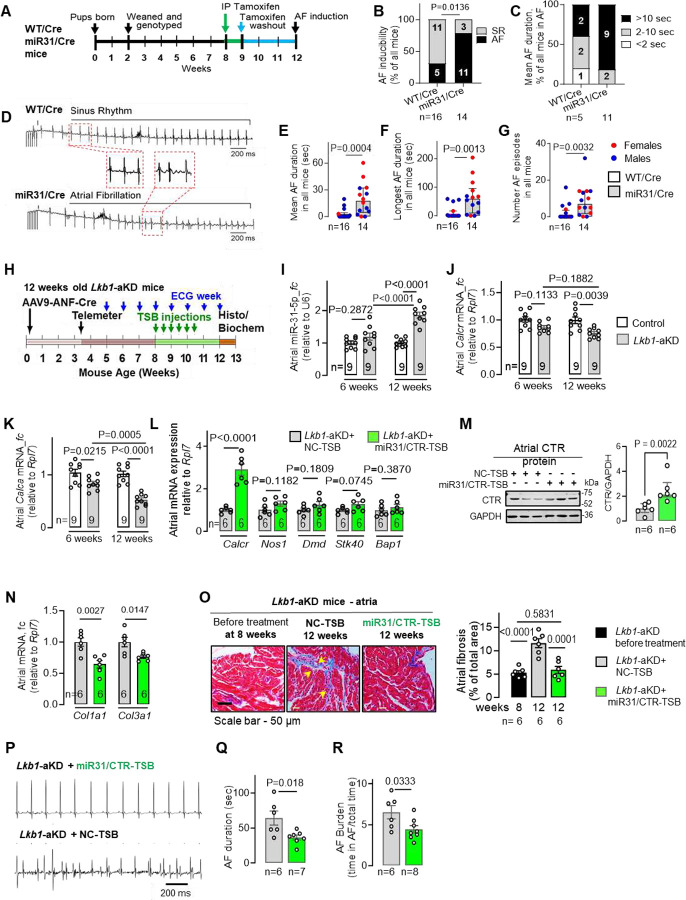 Figure 4
Figure 4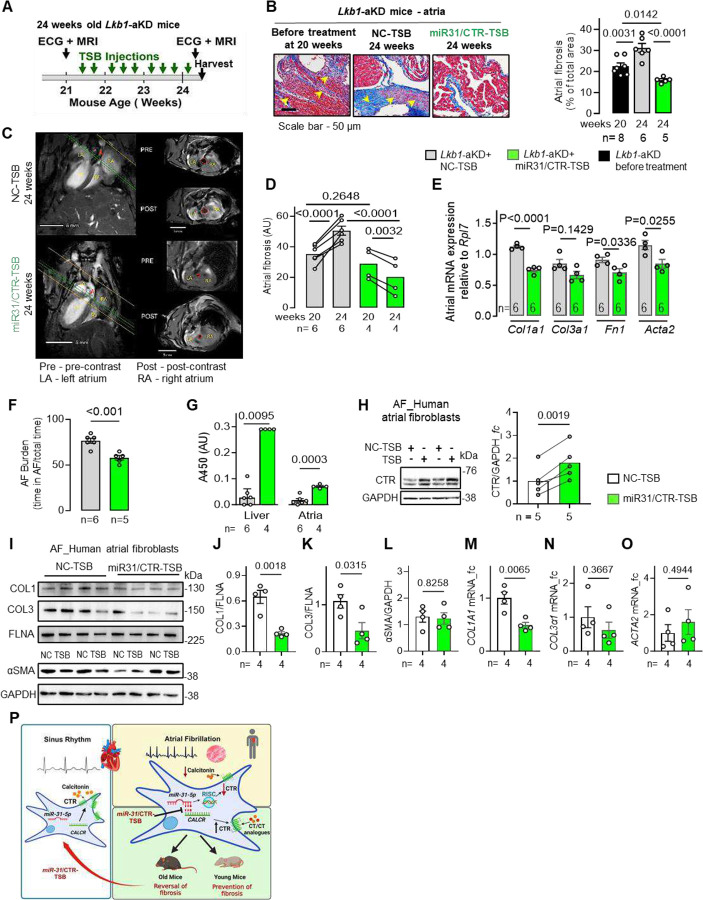 Figure 5
Figure 5Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsCardiac Fibrosis and Remodeling · MicroRNA in disease regulation · Atrial Fibrillation Management and Outcomes
INTRODUCTION
Atrial fibrillation (AF), the most common cardiac arrhythmia in humans, is a growing epidemic with an estimated incidence predicted to double by 2060, largely due to ageing. AF is a significant contributor to global mortality and morbidity, mainly due to stroke (AF alone is responsible for ~30–40% of all strokes) and heart failure (1, 2). Treatment of AF remains a major clinical challenge because of the underlying myocardial electrical and structural remodelling (3, 4). Electrical remodelling, caused by changes in ion channel expression/function, is largely reversible with maintenance of a normal sinus rhythm (SR) with the currently available anti-arrhythmic drugs, cardioversion and catheter ablation. By contrast, structural remodelling, hallmarked by atrial fibrosis, has not been successfully targeted by currently available therapies and is of particular concern in the clinic, as atrial fibrosis hampers AF treatment and worsens prognosis (5, 6). Thus, there is a quest to identify players in atrial fibrosis and cardiac remodelling to allow the development of more effective therapeutics for patients with this serious condition.^7^
Characterised by increased extracellular matrix (ECM) deposition without a significant loss of atrial cardiomyocytes (ACMs), atrial fibrosis increases as AF progresses (4) and is a key feature of cardiac remodelling in AF (8). The degree of atrial fibrosis negatively correlates with the success of one of the most successful therapeutic options, catheter ablation (9, 10). Despite extensive prior work, which uncovered key signalling pathways like those linked to transforming growth factor β1 (TGFβ1), galectin-3 and the renin-angiotensin-aldosterone system in atrial fibrosis, this insight has failed to yield clinically effective therapeutics to reverse atrial fibrosis. Therefore, there remains a major need to identify new players in atrial remodelling that can be targeted to generate more effective therapeutics for patients with AF (7, 9 10).
Atrial fibrosis is facilitated by complex interactions between cellular and neurohormonal mediators (11, 12), including a recently discovered cardioendocrine calcitonin (CT) system (13). This axis potently regulates atrial fibrogenesis and arrhythmia and is dysfunctional in the atria of patients with advanced atrial structural and electrical remodelling, such as in persistent-AF (persAF) (13). Cardiac CT produced by human ACMs controls proliferation, migration, and ECM production of atrial cardiofibroblasts (ACFs) (13). Disrupted signalling along this paracrine axis promotes collagen deposition, atrial fibrosis, and AF in vivo in mice. Patients with persAF have six-fold lower atrial CT production, reduced CTR expression and increased receptor internalization, facilitating enhanced collagen deposition by ACFs. Thus, restoring cardiac CT-CTR signalling represents an attractive new strategy to overcome major challenges in controlling cardiac remodelling in AF. While deficient CT can be replenished with exogenous CT-analogues (e.g. miacalcin, salmon CT) already in clinical use (e.g. for Paget’s disease, hypercalcaemia, osteoporosis), strategies to restore atrial CTR expression in persAF, a prerequisite for CT-mediated atrial anti-fibrotic and anti-arrhythmic effects, do not exist.
Given the reduced CTR protein in the absence of changes in CTR mRNA in ACFs from patients with persAF (13), it is plausible that microRNAs (miRs) may be involved in repressing CTR signalling. MiRs are endogenous small (19–27 nucleotides) non-coding RNAs which, via binding to a (partially) complementary sequence of 3’-untranslated mRNA region (3’UTR, or less commonly 5’UTR/CDs regions), mediate gene silencing by accelerating mRNA degradation or repressing translation (14). Previous work reported a number of miRs implicated in AF (15) including atrial-enriched miR-31–5p (16, 17), which, alongside three other miRs, is strongly predicted to bind the 3’UTR of the human CTR gene (CALCR). MiR-31–5p is implicated in ACM electrical remodelling in patients with persAF (17), in ageing (the strongest risk factor for AF (18)) and age-/senescence-related processes like cellular senescence, cancer and osteoporosis (19–21) (a condition associated with suppressed CT signalling). Our pilot work demonstrated that ACFs from patients with AF, which are characterised by supressed CTR-protein levels, produce twice the amount of miR-31–5p. Therefore, this study aimed to explore (i) the role of miR-31–5p as an upstream regulator of CTR signalling in remodelled atrial myocardium from patients with persAF, and (ii) the therapeutic potential of targeting the miR-31–5p/CTR mRNA interaction to prevent and/or reverse atrial fibrosis and arrhythmia.
METHODS
Patient cohorts.
Investigations using human samples were approved by the South Central-Berkshire B Research Ethics Committee (REF: 18/SC/0404). All patients gave informed written consent. A total of 136 patients were included in the study; all patients underwent cardiac surgery (coronary artery bypass or valve repair/replacement) in the John Radcliffe hospital, Oxford. Right and left atrial biopsies were collected during cardiac surgery, mostly before or shortly after (for LAA samples) cardiopulmonary bypass and immediately processed for cell isolation (described below) or snap-frozen until use in subsequent experiments (e.g., mRNA expression and immunoblotting).
Animals.
Animal breeding, handling and experimental work were carried out in two centers, Baylor College of Medicine (Houston, USA) and the University of Oxford (UK) according to the local home office standards. All animal work was performed in accordance with the UK Home Office Animals (Scientific Procedures) Act 1986 incorporating Directive 2010/63/EU of the European Parliament, and in accordance with NIH guidelines (USA). Animals were kept in pathogen-free cages with controlled temperature and humidity. The total number of mice used in all animal studies was 175 consisting of 66 of Lkb1-aKD and 109 of miR-31/Cre mice. Animals of both sexes were used in the experiments.
Generation of Lkb1-aKD mice.
The Lkb1-aKD mice were generated as previously described (13, 22). Briefly, AAV9-ANF-Cre was injected into 2–5 days old Lkb1^FL/FL^ pups (5×10^11^ GC, s.c. injections) generating atrial specific knockdown of Lkb1 (Lkb1-aKD). Based on our previous studies (13, 22), these mice develop ectopic activities from week 5–6 that can be detected by surface ECGs. A 15-minute surface ECG was performed weekly to confirm ectopic activity and/or the onset of AF (from week 8 onwards). These mice were then treated with NC- or miR-31/CT-TSB either at 8 or 20 weeks (for the MRI cohort). Please see Supplementary Materials for more details.
Generation of miR-31/Cre mice.
The miR-31/Cre mice were generated by PhiC31 integrase mediated cassette exchange in mouse embryonic stem cells to integrate a Cre recombinase-activatable Pre miR-31 expression cassette at the Gt(ROSA26)Sor locus. Please see Supplementary Materials for more details.
Isolation and culture of primary human atrial cardiofibroblasts (ACFs).
Human ACFs were isolated and cultured from atrial biopsies obtained from patients who underwent cardiac surgery, as previously described (13). Tissue biopsies were cut into small (2–3 mm^3^) pieces and repeatedly digested using 4 mg/ml collagenase II and trypsin (0.0625%), as previously described (13). Cells were washed twice with sterile phosphate-buffered saline (PBS) and plated onto 6-well plates in FBM-3 medium (#CC-3131, Lonza) containing 10% fetal bovine serum (FBS) and a supplement pack (#CC-4525, Lonza) and kept in a humidified atmosphere at 37 °C and 5% CO_2_. Please see Supplementary Materials for more details.
Sources of other human cells.
In addition to the human primary cells, we also used HEK293 cells (#CRL-1573, ATCC) cultured and maintained according to the manufacturer’s instruction.
Transfection of primary human atrial cardiofibroblasts (ACFs).
Human ACFs were transfected with miR-31–5p mimic, miR-31–5p inhibitor, or respective negative controls (NC-mimic, NC-inhibitor), or with miR-31/CTR-TSB and NC-TSB as previously described (16). Specifically, blockade of miR-31–5p/CTR interaction was achieved with 25 or 50 nM of antisense miRCURY LNA^™^ (locked nucleic acid) miRNA Power Target Site Blocker (TSB; #339199, Qiagen) GGGCAAGAGGTAATCT, or miRCURY antisense LNA^™^ Power Target Site Blocker negative control A (NC-TSB, #339199, Qiagen) ACGTCTATACGCCC*A; LNA is not shown, as this information is proprietary. Cells were transfected using Lipofectamine RNAiMAX transfection reagent (#13778030, Invitrogen) in antibiotic-deprived FBM-3 medium containing 2 % FBS (Lonza).
Inhibition or overexpression of miR-31–5p was achieved by miRIDIAN miR-31–5p inhibitor (or NC-inhibitor) or mimic (or NC-mimic), respectively (all from Dharmacon: #IH-300507–06, IN-001005–01, C-300507–05 and CN-001000–01), using lipofectamine RNAiMAX transfection reagent (#13778030, Invitrogen) diluted in Opti-MEM medium (#31985062, Gibco), as described (16.) Efficient knockdown and overexpression was confirmed by real-time qPCR and visually by using the miRIDIAN Dy547-labeled transfection control (for experiments with miR-31–5p mimic or inhibitor). Please see Supplementary Materials for more details.
Western blot.
Immunoblotting in human and murine atrial homogenates and in human ACFs was performed as described previously (13). Briefly, tissue or cell samples were lysed in RIPA buffer or CelLytic M cell lysis reagent, electrophoresis was performed using pre-cast 4–12% NuPAGE gels (#NP0335BOX, Invitrogen) and MOPS SDS running buffer (#NP0001, Invitrogen) at 150V. Proteins were transferred onto 0.2 μm nitrocellulose membrane (#1620112, Bio-Rad) using transfer buffer (#NP0006, Invitrogen). Immunodetection of the primary antibodies (Table 2, Supplementary Information) was performed using anti-IgG HRP-conjugated secondary antibodies (Table 2, Supplementary Information). Membranes were developed using a ChemiDoc System (BioRad, UK) and analysed with ImageJ software (version 1.52a) or Image Lab 6.1 (Bio-Rad). For stripping, nitrocellulose membranes were submerged in Restore PLUS Striping buffer (#46430, Thermo Scientific). The target protein and a loading control were always run on the same membrane.
Colorimetric assays.
Quantification of total secreted collagen in the cell culture supernatant was performed using a Sirius Red collagen detection kit (#9049, Chondrex) as previously described (13). The amount of total collagen in murine atrial lysates was quantified by colorimetric detection of hydroxyproline using a Quickzyme total collagen assay kit (#QZBTOTCOL1, lot 0795, QuickZyme Biosciences).
Scratch wound migration assay.
Human ACFs migration was determined using in vitro scratch wound assays as previously described (13). Scratch wound assay was performed on confluent monolayers of cells (following 24-hour transfection with miR-31–5p mimic, miR-31–5p inhibitor, or respective negative controls) using chambers with 2 well silicone insert with a defined cell-free gap (#80206, Ibidi). Please see Supplementary Materials for more details.
Scar-in-a-jar assay.
Collagen 1 accumulation by fibroblasts was assessed using a scar-in-a-jar assay as in our previous work (13). Please see Supplementary Materials for more details.
Assessment of cell proliferation.
Cell proliferation at a single time point was assessed using BrDU (5-Bromo-2’-Deoxyuridine) DNA-binding probe (#QIA58, Sigma-Aldrich) according to the manufacturer’s instructions and as previously described (13). Please see Supplementary Materials for more details.
Assessment of gene expression with Real-Time Quantitative Polymerase Chain Reaction (qPCR).
As previously described (13, 16), atrial tissue samples were homogenised using an electric homogeniser Polytron with an appropriate lysis buffer. Total RNA was extracted with Nucleospin RNA II kit (#740955.250, Macherey-Nagel) following manufacturer’s instruction. Complementary DNA (cDNA) was synthesised from 250 ng of RNA with high-capacity cDNA reverse transcription kit (#4368814, Thermofisher Scientific) according to the manufacturer’s protocol. Total RNA from human ACFs was extracted with mirVana miRNA isolation kit (#AM1561, Invitrogen). cDNA was synthesised from 100 ng RNA using QuantiTect Rev. Transcription Kit (#205313, Qiagen). For miR-31–5p/3p reverse transcription (RT), the TaqMan MicroRNA Reverse Transcription Kit (#4366596, Applied Biosystems) and TaqMan MicroRNA assay RT primers were used. Please see Supplementary Materials for more details.
RNA-binding protein immunoprecipitation (RIP) with Ago2 in human ACFs.
Human ACFs from patients with persAF were transfected with miR-31/CTR-TSB or NC-TSB (25 nM) for 24 hours. The RIP assay was performed using the Magna RIP kit (#17–700, Sigma-Aldrich) as previously described (16). In brief, cells were washed in ice-cold PBS (two times) and lysed in 100 μl complete RIP lysis buffer. Magnetic beads were conjugated with 5 μg of anti-Ago2 antibody (#ab32381, Abcam) and subsequently incubated with the RIP lysates overnight at 4°C with rotation. Immunoprecipitates were purified and kept overnight at −80°C to precipitate the RNA and resuspended in RNase-free water. RNAs were subjected to RT-qPCR to measure the CALCR mRNA and miR-31 levels. CALCR gene expression was normalised to miR-31 and quantified using the comparative threshold cycle method (2^−ΔCt^). Please see Supplementary Materials for more details.
Transcription inhibition with actinomycin D.
Human ACFs from patients in SR were transfected with 25 nM of miRIDIAN miR-31–5p mimic or NC-mimic for 24 hours and then treated with 5 μg/ml actinomycin D (#A9415, Sigma-Aldrich) for ~8.5 hours. Levels of CALCR (qPCR) are shown as percentage of the respective mRNA (2^−ΔCt^) at 0 hour, normalising to GAPDH as the housekeeping gene. Please see Supplementary Materials for more details.
Dual-Glo firefly luciferase reporter assay in HEK293 cells.
The reporter construct was generated using the pmirGLO Dual-Luciferase miRNA Target Expression Vector (#E1330, Promega) containing the whole length (1869 bp) of CALCR 3′UTR. Please see Supplementary Materials for more details.
Histological assessment of cardiac fibrosis with Masson’s trichrome staining in mice.
Staining for collagen was performed using Masson’s trichrome staining as described before (13, 22). All experiments and analysis were performed blinded. Please see Supplementary Materials for more details.
Transthoracic echocardiography.
Cardiac functions of the Lkb1-aKD, miR-31/Cre mice and control mice were assessed with transthoracic echocardiography. Please see Supplementary Materials for more details.
In vivo transesophageal burst pacing in miR-31/Cre and WT/Cre mice.
AF inducibility in miR-31/Cre and WT/Cre mice was assessed with transesophageal burst pacing as described previously (16, 23). Please see Supplementary Materials for more details.
Telemetry studies.
Telemetry studies were performed as described previously (26). Please see Supplementary Materials for more details.
MRI in mice.
Mice underwent MRI before miR-31/CTR-TSB or NC-TSB treatment and 3.5 weeks later. All MRI imaging data were acquired using a Bruker Biospec 9.4 T, 20 cm bore, AV NEO/Paravision 360 at the Small Animal Imaging Facility at Texas Children Hospital. Please see Supplementary Materials for more details.
In vivo miR-31/CTR-TSB administration in mice.
Blockade of the miR-31/CTR interaction was achieved with antisense miRCURY LNA miRNA Power TSB (#339202, Qiagen) GGGCAAGAGGTAATC*T, or NC-TSB (#339202, Qiagen); LNA is not shown, as this information is proprietary. Please see Supplementary Materials for more details.
A sandwich hybridization assay of LNA-TSB.
A sandwich hybridisation assay was performed using a biotinylated LNA-capture probe complementary to half of the LNA-miR-31/CTR-TSB to capture the LNA-TSB on streptavidin-coated magnetic beads. Please see Supplementary Materials for more details.
RNA sequencing: sample processing and analysis.
Total RNA was extracted from hACF cell cultures using mirVana miRNA Isolation Kit (Thermo: AM1560) as above. Please see Supplementary Materials for more details.
Statistical analysis.
The data distribution was tested by Kolmogorov-Smirnov or Shapiro-Wilk tests. The unpaired or paired Student’s t test was used in two-group comparisons of the normally distributed data. Multiple groups of normally distributed data of similar variance were compared by one-way or two-way ordinary or repeated measures ANOVA with Holm-Sidak’s or Sidak’s correction. When the normality assumption was not met the data were analysed by Mann-Whitney or Wilcoxon test - for independent or paired comparisons between 2 groups, respectively – or Kruskal-Wallis with Dunn’s correction - for multiple group comparisons. Categorical variables were compared by Fisher’s exact test. All statistical analysis was performed using GraphPad Prism v7, v6.04, v9.0 and v10 software. A value of P < 0.05 was considered statistically significant. Pearson’s correlation test was used to test correlation between 2 normally distributed variables.
Data availability:
All data generated or analysed during this study are included in the article. The RNAseq data is deposited REF: GSE294333 at https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE294333
RESULTS
MicroRNA-31–5p regulates CTR abundance in human ACFs.
Using published reports (table S1) of the miRs altered in atrial tissue or cells in AF, we identified, using in silico mirDIP analysis at https://ophid.utoronto.ca/mirDIP/, four miRs (hsa-miR-24–3p, -* 30d-5p*, −31–5p and 34a-5p), which are strongly predicted to bind CTR-mRNA (CALCR) 3’UTR (fig. S1A). A reporter assay showed no binding between CALCR-3’UTR and hsa-miR-24–3p, −30d-5p, or −34a-5p in HEK293 cells co-transfected with pmiRGLO luciferase/renilla-expressing plasmid containing full-length CALCR-3’UTR and the respective miR mimic/inhibitor (vs mimic/inhibitor-non-targeting negative controls [NC]) (fig. S1B–H). By contrast, the reporter assay showed ~60% reduction of the luciferase/renilla ratio (P=0.001) in cells transfected with miR-31–5p mimic (vs NC-mimic) that was fully reversed by miR-31–5p inhibition, indicating that miR-31–5p binds the CALCR-3’UTR (fig. 1A–C). While upregulation of miR-31–5p has been reported in human ACMs and noted to be relevant to electrical remodeling in AF (16, 17), its role in ACFs and its potential contributions to AF fibrogenesis is unknown. We found that in patients (table S2) with persAF, compared to SR controls, ACFs from both right (RAA; P=0.003) and left (LAA; P=0.008) atrial appendages had 2.7- and 2.5-fold increased miR-31–5p expression respectively (fig. 1D, E); notably, miR-31–5p expression was barely detectable in the left ventricular myocardium (fig. S1I). Moreover, miR-31–5p levels negatively correlated with CALCR expression in these ACFs (RAA: P=0.029; LAA: P=0.049; fig. 1F, G). To test the effect of miR-31–5p on CTR gene and protein expression in native ACFs, ACFs were isolated from SR and persAF patients were transfected with miR-31–5p mimic (or NC-mimic) and miR-31–5p inhibitor (or NC-inhibitor) respectively. Overexpression of miR-31–5p (fig. 1H) halved CTR-mRNA (P=0.0028) and protein (P=0.0312) (fig. 1I, J), while miR-31–5p inhibition (fig. 1K) increased CTR-mRNA by ~30% (P=0.0183) and nearly doubled CTR protein (P=0.017) vs NC-treated ACFs (fig. 1L, M). These results indicate that miR-31–5p inhibition is sufficient to restore the decreased ACF-CTR expression previously observed in persAF (13).
To confirm the endogenous interaction between miR-31–5p and CALCR (i.e., occupancy of miR-31–5p on CALCR-3’UTR) we assessed, with previously validated RNA-binding protein immunoprecipitation (RIP) assay and qPCR (16), the loading of both targets into the RNA-Induced-Silencing-Complex (RISC) in AF-ACFs. While both CALCR and *miR-31-*5P were present in Ago2-RISC pulldowns (fig. 1N, O), a selective blockade of the miR-31–5p/CALCR interaction with LNA-power-Target-Site-Blocker (miR-31/CTR-TSB) decreased CALCR, but not miR-31–5p, RISC content by ~60% (P=0.0334) (fig. 1N, O), further confirming an endogenous miR-31–5p/CALCR interaction in human AF-ACFs. To elucidate the mechanism (enhanced mRNA degradation or translational silencing) by which miR-31–5p regulates CALCR, we compared the effects of miR-31–5p mimic (or NC-mimic) (fig. 1P) on CALCR levels in human ACFs (hACFs) pre-treated with 5 μg/ml actinomycin D, a potent, non-selective transcription inhibitor (17); CALCR diminished faster in the presence of miR-31-5p mimic (vs NC-mimic) (P=0.0114), pointing towards enhanced mRNA degradation as the predominant mechanism by which miR-31–5p regulates CTR expression in hACFs (fig. 1Q).
MicroRNA-31–5p regulates CTR expression in atrial fibroblasts in mice.
Similar to humans, the mouse CTR (Calcr) mRNA also contains a conserved miR-31–5p binding site in its 3’UTR (fig. 2A). To test the effect of miR-31–5p /Calcr binding in vivo, we generated a tamoxifen-inducible fibroblast-targeted miR-31 transgenic mouse model (miR31/Cre) using *Col1a2-*Cre-ER(T) transgene (fig. 2B, C; fig. S2, S3A). A qPCR confirmed successful overexpression of murine miR-31–5p in atrial and ear (a positive fibroblast-enriched control) tissue lysates (fig. 2D, E) in miR31/Cre mice vs control WT/Cre, or other relevant genotypes (WT/WT and miR31/WT), in both female and male animals. The miR-31–3p strand, which does not have a binding site for CTR-mRNA-3’UTR, was also (though to a lesser extent) upregulated (by ~30%, fig. S3B compared to ~2-fold for miR-31–5p, fig. 2F), in murine ACFs. Analysis of miR-31–5p expression in isolated atrial and ventricular cardiofibroblasts (CFs) and cardiomyocytes (CMs) showed a 2.5- and 1.6-fold increased expression in atrial-CFs (P=0.003) and ventricular-CFs (VCFs), though it did not reach statistical significance in VCFs (P=0.062), with unchanged levels in atrial-CMs (P>0.999) and ventricular-CMs (P=0.700) (fig. 2F, G). These results indicate the successful overexpression of the miR-31 transgene CFs. Neither miR-31-transgene nor tamoxifen administration affected ventricular function in mice in either genotype in males or females (fig. 2H–M, S4A-H; table S3, S4, S5). Similar to hACFs, murine miR-31 overexpression significantly decreased atrial CTR protein and atrial fibroblast mRNA (fig. 2N, S5A) compared to the control WT/Cre animals. This was associated with unchanged expression of the CTR ligand calcitonin (CT) transcript (Calca) expression in murine ACMs (fig. 2O).
Effect of miR-31–5p on hACF functions.
To evaluate the functional responses of hACFs to miR-31 overexpression or inhibition, we performed in vitro studies in these cells. ECM collagen-1 accumulation (assessed by scar-in-a-jar assay) was ~30% increased (P=0.0118) by 72-hour overexpression of miR-31–5p mimic in SR-hACFs (fig. 3A, B). By contrast, inhibition of miR-31–5p with miR-31–5p inhibitor halved collagen-1 content in ACFs from patients with persAF (fig. 3C, D). Similar effects were obtained in the presence of TGFβ1 stimulation (5 ng/ml, fig. S4I, J). MiR-31–5p mimic had no effect on fibronectin protein in hACFs (fig. 3A–D) – possibly due to a maximal activation of cell responses in persAF. Expression of α-smooth muscle actin (αSMA) increased 2-fold (P=0.0339) in miR-31–5p overexpressing SR-hACFs, while it was halved (P=0.0346) by miR-31–5p inhibition in AF-hACFs (fig. 3E, F). While overexpression of miR-31–5p did not alter transcript expression of COL1, ACTA2 and FN1, miR-31–5p inhibition reduced ACTA2 (P=0.0038) with a trend towards a reduction for COL1 (P=0.056) and unchanged FN1 in hACFs (fig. 3G, H). Cell proliferation was 50% higher (P=0.0176) in miR-31–5p mimic SR-hACFs (fig. 3I) and reduced by ~29% (P=0.0311) by miR-31–5p inhibition in AF-ACFs (fig. 3J). However, it remained unchanged in TGFβ1-activated hACFs in both miR-31–5p mimic or inhibitor treated groups (fig. S4K, L), likely attributable to maximal ACF activation and proliferation response to TGFβ1. Neither miR-31–5p mimic (vs NC-mimic; fig. 3K) nor miR-31–5p inhibitor (vs NC-inhibitor; fig. 3L) affected the migration of hACF. These results suggest that overexpression of miR-31–5p in vitro is sufficient to activate some key fibrotic responses (collagen-1 and αSMA production, and cell proliferation) in hACFs which, in the presence of persAF, are significantly suppressed by miR-31–5p inhibition.
Upregulation of miR-31 in fibroblasts promotes structural and arrhythmogenic substrate in vivo.
To test whether the atrial fibrotic remodelling in persAF caused by the loss of CTR is secondary to miR-31 upregulation, we used the fibroblast-specific miR-31-overexpressing mice (fig. 2). These mice displayed a doubled (P=0.0025) amount of atrial (but not ventricular) fibrosis assessed by Masson’s trichrome staining (fig. 3M, N) and hydroxyproline assay (P=0.028) (fig. 3O, P) compared to the WT/Cre controls. These alterations were accompanied by ~40% increase in atrial protein levels of collagen-1 (P=0.016), collagen-3 (P=0.006) and αSMA (P=0.049) (fig. S5A, B, C), while mRNA was unchanged for collagen-1, increased for collagen-3 and reduced for αSMA (fig. S5D-F). Atrial fibronectin protein remained unchanged (despite decreased its mRNA; P=0.0429; fig. S5A, G). By contrast, left ventricular collagen-1, collagen-3 and fibronectin proteins remained unaltered, while αSMA increased (fig. S5H, I). Overexpression of miR-31 also caused a reduction in the mRNA (fig. S5J-M) and, in some cases, protein (with the exception of BAP1 - remained unchanged, and TNS1 - increased) expression in the atria of previously validated miR-31-targets, FGF-1, BAP1, STK40 and TNS1 (fig. S5A, B). Increased atrial fibrosis in mice was associated with a 2-fold higher AF inducibility (P=0.0136) (fig. 4A, B), prolonged AF duration (fig. 4C–F) and more frequent AF episodes (fig. 4G). As miR-31 overexpression in mice did not differ between males and females and had no correlation with sex, and there were no sex-dependent differences in heart rate, ventricular function or AF duration/number of AF episodes, we did not analyse males and female results separately in the downstream experiments. Our findings suggest that a clinically relevant 2-fold upregulation of miR-31 in atrial fibroblasts (fig. 2F) is sufficient to promote the formation of an AF substrate in vivo.
MiR-31–5p alters the transcriptional programme of hACFs.
To assess whether miR-31–5p alters other potential target genes and the broader transcriptional programme of hACFs, control SR-ACFs were transfected with miR-31–5p mimic, and AF-ACFs with miR-31–5p inhibitor (or respective NC-controls), and submitted for bulk RNA-sequencing (RNAseq) (fig. S6). Principal component analysis (PCA) revealed clustering of different groups by treatment and a distinct pattern in differentially expressed genes (DEGs) between miR-31–5p mimic and inhibitor vs NC-mimic/NC-inhibitor groups (fig. S6A-D). While miR-31–5p overexpression significantly upregulated 1023 and downregulated 1009 genes, miR-31–5p inhibition upregulated 70 and downregulated 70 genes (fig. S6C, D; data S1). RNAseq and qPCR confirmed concordant changes in previously reported miR-31 gene targets, BAP1, PKN2 and STK40 (24, 25), in hACFs transfected with miR-31–5p mimic and inhibitor (fig. S6E-N). DEG pathway analysis found that miR-31–5p overexpression leads to the activation of microtubule movement/formation, as well as pathways involved in cilium organisation and assembly (fig. S6O), while inhibiting cell cycle and nuclear chromosome organisation and function (fig. S6P). These transcriptional changes may contribute to the miR-31-mediated fibrotic responses in AF-hACFs (26). By contrast, miR-31–5p inhibition in AF-hACFs alters genes involved in TGFβ, TNF and steroid hormone signalling (fig. S6Q). Notably, miR-31–5p mimic treatment elevated the expression of several fibrosis-relevant genes (e.g. BMP1, COL3, FAP, FN1, POSTN and THBS1; fig. S6R), the relevance of which should be explored in future work. MiR-31–5p inhibition was also associated with suppressed inflammation-related DEGs, CCL5, CCL26, TNFAIP6, IFI6 and HSPA6/7 (fig. S6S). The concordant changes between miR-31–5p and these DEGs suggest an indirect effect of miR-31–5p on them.
Selective disruption of miR-31–5p/CALCR interaction reverses atrial fibrosis and arrhythmia in vivo.
LNAs are modified nucleic acids with enhanced binding affinity for their target RNA (27). Using miRCURY LNA-miR-TSB in vivo to selectively block the mi-R31–5p/CALCR interaction, we tested causality between the miR-31–5p/CALCR interaction, atrial fibrosis, and AF development in the previously-validated Lkb1-aKD mouse model of spontaneous AF (fig. 4H) (13, 22). Similar to the changes observed in patients with persAF (13) (fig. 1B–E), the Lkb1-aKD mice displayed almost doubled levels of miR-31–5p (fig. 4I), and reduced (by ~30%) Calcr- and Calca-mRNA (fig. 4J, K) (vs Lkb1^FL/FL^ animals) at 12 (and for Calca as early as 6) weeks of age. Thus, Lkb1-aKD mice represent a clinically relevant model in the context of the AF-associated activation of miR-3–5p1/CTR signalling.
We initially injected 8-week-old Lkb1-aKD mice with miR-31/CTR-TSB or respective NC-TSB and monitored structural remodelling (atrial fibrosis) and AF burden/duration for 4 weeks. As 8-week-old mice do not exert established fibrosis and structural remodelling, they were used to test preventive value of the TSB treatment on structural remodelling and arrhythmogenesis. We observed that miR-31/CTR-TSB treatment rescued atrial CTR-mRNA (P=0.0001) and protein (P=0.0076) (fig. 4L, M) without changes in other previously validated targets of miR-31 (Nos1, dystrophin (Dmd), Stk40 and Bap1; fig. 4L), thus confirming the specificity of the TSB approach to disrupt the miR-31–5p/Calcr interaction. Compared to NC-TSB, the miR-31/CTR-TSB treatment suppressed expression of the atrial collagen-1 (by ~40%, P=0.0027) and collagen-3 (by ~25%, P=0.0147) (fig. 4N), prevented the development of atrial fibrosis (P=0.0001) assessed with Masson’s trichrome stain (fig. 4O), and reduced AF duration by ~45% (P=0.018) and burden by ~32% (P=0.0333) (fig. 4P–R). Therefore, selective blockade of miR-31/CALCR binding is sufficient to preclude atrial fibrogenesis and control atrial arrhythmogenesis in vivo in mice.
While the prevention of atrial fibrosis was achievable in younger mice by therapy beginning before the development of fibrosis, we next questioned whether selective blockade of miR-31-5p/CALCR binding could exert a therapeutic effect on established atrial fibrosis (commonly observed in persAF patients) and arrhythmia. Thus, we subjected older Lkb1-aKD mice (20.5 weeks of age, with ~26% baseline atrial fibrosis, similar to the level observed in persAF patients) (29) to a 3.5-week treatment with miR-31/CTR-TSB, or NC-TSB (25 mg/kg, i.p., thrice per week) (fig. 5A). Atrial arrhythmias and fibrosis were monitored at baseline (20-weeks) and post-TSB treatment (24-weeks) by ECG and MRI (using a collagen-1 binding Gadolinium-diethylenetriamine pentaacetic acid dye, EP3533), respectively (29). Baseline atrial fibrosis did not differ significantly between groups; however, it decreased by ~30% (compared to the pre-treatment 20-week fibrosis levels) post-TSB treatment (confirmed by Masson’s trichrome staining - fig. 5B), while in the NC-TSB control group atrial fibrosis progressed by ~46% above the 20-week baseline fibrosis (P<0.0001) (Fig. 5C, D). The miR-31/CTR-TSB-mediated reversal of atrial fibrosis was accompanied by a reduction in atrial mRNA expression of collagen-1 (by 33%, P=0.0001), fibronectin (by 22%, P=0.0336) and αSMA (by 26%, P=0.0255) with unchanged collagen-3 (fig. 5E) in TSB-treated (vs NC-TSB) mice. ECG revealed reduced AF burden (by ~16%; P= 0.033) in miR-31/CTR-TSB-treated mice (fig. 5F). A sandwich hybridization assay with a biotinylated LNA-capture probe showed efficient delivery of the miR-31/CTR-TSB to the atrial myocardial, with 4.2- and 8.1-fold increase (vs NC) in the atria and liver respectively (fig. 5G) (30). The miR-31/CTR-TSB treatment had no effect on blood pressure, heart rate, or cardiac contractility (table S5). In hACFs, the TSB-mediated selective inactivation of miR-31/CALCR binding approximately doubled CTR protein (P=0.0125) (fig. 5H) and suppressed mRNA and protein levels of collagen-1 (by ~68%; P=0.0018 and 51%; P=0.0065 respectively), reduced collagen-3 protein (but not mRNA) by 56% (P=0.0315) without changes in αSMA expression (fig. 5I–O). These observations confirm that the actions of miR-31/CTR-TSB in the in vivo mouse model are relevant to human atrium, and they demonstrate that selective deactivation of the miR-31/CTR signalling axis with miR-31/CTR-TSB reverses the established atrial fibrosis in vivo.
DISCUSSION
Here, we identified miR-31–5p as a novel, upstream regulator of CTR signalling, whereby miR-31–5p binds the CTR-3’UTR to enhance its degradation/reduce translation and suppress CTR expression in human atrial fibroblasts, and promotes atrial fibrosis and arrhythmogenesis in vivo. Activation of this signalling cascade in the presence of heart disease, such as AF, or in a *miR-31-*Tg mouse model, induces fibrotic fibroblast activity leading to excessive atrial fibrogenesis and increased arrhythmia susceptibility. Patients with persAF, characterised by established atrial fibrosis and decreased CTR expression (13), have doubled miR-31–5p levels and increased miR-31–5p/CALCR interaction in ACFs. Animal studies showed that selective disruption of this signalling axis, with a miR-31–5p/CTR-TSB, was sufficient to prevent new (in young mice) and reverse advanced (in older mice) atrial fibrosis and arrhythmogenesis in vivo (fig. 5P).
A recently discovered endogenous, cardio-endocrine calcitonin signalling system in human atrial myocardium controls atrial fibrosis and arrhythmogenesis (13). Impaired CT-CTR signalling, as in patients with persAF, promotes atrial fibrogenesis and the formation of an arrhythmogenic substrate. While patients with persAF have suppressed CT production and CTR expression/internalization, they are unlikely to benefit from clinically approved CT-analogues unless CTR expression and function is restored. Furthermore, using CT-analogues is likely to further repress the already diminished cellular CTR surface expression as a result of ligand-induced and pre-existing (13) receptor internalisation and degradation, commonly observed with G protein-coupled receptors (31). Thus, in the light of the therapeutic potential of this signaling system for atrial fibrosis and arrhythmia, we searched for upstream mechanisms regulating CTR abundance in hACFs. We found that both right and left ACFs from patients with persAF have doubled levels of miR-31–5p (associated with ageing, and shown to play a role in ischemic and coronary heart diseases) (32, 33) that negatively correlated with CTR-mRNA (CALCR) expression, pointing towards a potential regulatory role for miR-31–5p on CALCR abundance. This was supported by the results of the reporter assay, which demonstrated changes in CTR gene/protein in hACFs after activation or suppression miR-31–5p, and by the confirmation of endogenous miR-31–5p/CTR-mRNA binding by RISC-pulldown in human AF-ACFs. Both miR-31–5p inhibition and selective disruption of miR-31–5p/CTR binding with TSB in vitro in human ACFs was sufficient, through restoring CTR expression, to repress some key fibrotic responses of ACFs, e.g., collagen production and proliferation.
Previous studies suggest transcriptional regulation of CTR expression (34). However, CTR mRNA expression is unchanged in AF patients (13), so transcriptional alterations are unlikely to play a major role in AF-hACFs in the intact human heart, despite miR-31–5p-mediated changes in CTR-mRNA expression in the in vitro studies using supra-physiological miR-31–5p levels. This discrepancy are likely due to a more complex endogenous environment and potential compensatory feedback in the endogenous myocardial environment of AF-patients.
While we found that miR-31–5p regulates CALCR abundance, it is common for a single gene to be regulated by multiple miRs (35); hence, CTR abundance in AF-ACFs might be controlled by other miRs. We tested the potential role of three other miRs previously documented to play a role in persAF that had a high predicted score for binding to CTR-3’UTR, but found no regulatory effects of those miRs on CTR. Notably, the logistic regression analysis showed that miR-31–5p was significantly associated with a higher risk of AF (Table S6, OD = 3.02, P-value = 0.025), suggesting that the upregulation of miR-31 in AF is not influenced by other confounding factors, highlighting its specific contribution to atrial fibrosis and AF pathogenesis.
In vivo studies confirmed the miR-31–5p mediated regulation of CTR in ffibroblast-targeted miR-31-overexpressing mice with a clinically relevant~2.5-fold (similar to that observed in patients with persAF) miR-31–5p upregulation. These mice had raised expression of both miR-31–5p and −3p, however, as latter has no binding site for CTR-3’UTR it was not studied further. Furthermore, given that a selective blockade of miR31–5p/CTR interaction with TSB successfully reverses atrial fibrosis and arrhythmogenesis indicates that this treatment is sufficient to preclude effects potentially mediated by actions via one of the other miR-31–3p targets. Notably, the changes observed in these mice were primarily driven by miR-31–5p produced by atrial (not ventricular) CFs, as ventricular-ACF fibrotic markers and LV-function did not differ between groups. Consistent with previous reports (16, 17), we observed greater levels of miR-31–5p in the atria, while it was barely detectable in ventricular myocardium (10-fold increase in atrial-CFs vs ventricular-CFs). Accordingly, we found that fibroblast-targeted miR-31–5p overexpression in mice caused an increase in atrial fibrosis associated with a significant prolongation of arrhythmic episodes and increased AF susceptibility. These changes seem to be independent of sex (we observed no differences between males and females), and developed in the absence of left atrial dilatation or changes in left ventricular systolic or diastolic function in miR-31/Cre mice. Therefore, a fibroblast-selective miR-31 expression increase, comparable to that seen in patients with persAF, is alone sufficient to promote structural, fibrotic remodelling that translates into enhanced arrhythmia-maintaining substrate and vulnerability to AF. Notably, miR-31–5p has also been shown to regulate the electrical properties of atrial-CMs in persAF (16, 17); therefore, it is plausible that miR-31–5p (via distinct gene targets) represents a common master-driver of electrical and structural remodelling of the atrial myocardium in AF.
To test the relevance of the miR-31–5p/CTR-mRNA interaction to atrial remodelling, we utilized a previously characterised Lkb1-aKD mouse model of spontaneous AF (22). These mice (similar to AF patients) have 2-fold upregulated miR-31–5p and reduced CTR expression and, thus represent a clinically relevant (in terms of miR-31-5p/CTR signalling) model. Selective blockade of the miR-31–5p/CALCR interaction in mouse atria with miRCURY LNA (25) miRNA power miR-31/CTR-TSB, which was successfully delivered to the atrial myocardium, specifically (without altering the expression of other miR-31 targets) increased atrial CTR expression, confirming an endogenous miR-31–5p/CTR interaction in vivo. Furthermore, 3-week TSB therapy in young (8-week-old) mice fully prevented the development of atrial fibrosis and significantly reduced both AF burden and duration. Although young mice displayed an atrial fibrotic burden (~6% of total area) comparable to that of non-transgenic age-matched mice, this does not reflect the typical clinical presentation of patients with established structural remodelling, like in persAF, who typically exhibit advanced (~22%) atrial fibrosis (28). Thus, we administered TSB (with higher frequency treatment regimen) in older (20-week-old) mice with established (~26%) atrial fibrosis. Pre- and post-treatment (at 20 and 24 weeks) MRI and histological assessment with Masson’s trichrome staining showed a reversal (by ~30% below the 20-week baseline) of atrial fibrosis in Lkb1-aKD mice in vivo. As in mice, miR-31/CTR-TSB treatment of human AF-ACFs suppressed production of some key fibrotic markers (e.g., collagen-1/−3). Hence, miR-31/CTR-TSB administration might offer a new approach to reverse advanced structural remodelling in the atrial myocardium, such as that encountered in patients with persAF. In a canine model of HF-induced atrial fibrosis and AF, complete reversal of HF does not lead to reversal of fibrosis and the AF substrate (36), so active therapy is likely needed to decrease already-established fibrosis. Given that miR-31 upregulation in persAF is atrial specific (16) such therapy may help to circumvent off-target effects on ventricular myocardium, a common limitation of many current AF therapies (37). Furthermore, use of selective miR-mRNA-TSB agents may help to evade the concerns of off-target effects presented by global miR inhibition (37), e.g., changes in multiple (including physiologically important) miR-31 targets identified by RNAseq. Encouragingly, miR-31/CTR-TSB administration had no effect on previously identified miR-31 targets (e.g., Nos1, Dmd, Stk40, Bap1), confirming the high selectivity of the miR-31/CTR-TSB approach in hACFs and mouse atrial myocardium. Strikingly, 3-week TSB pulse-therapy was effective in not only preventing fibrosis from developing, but also reversed (by 30%) advanced atrial fibrosis in vivo in older Lkb1-aKD mice.
Given that ageing-/senescence-related processes (18–20), as well as ischemic (21) or coronary heart disease (24) can lead to miR-31 upregulation, it is conceivable that miR-31 upregulation in AF might be secondary to the underlying cardiac pathology, or ageing, rather than driven by AF itself. However, the latter cannot be excluded, as miR-31 rises in Lkb1-aKD mice who display aging-related remodelling localised to atrial (but not ventricular) myocardium. Intriguingly, both upregulation of miR-31–5p and impaired CT/CTR signaling are also linked to the age-associated processes in bones and joints, osteoporosis and osteoarthritis (19, 38–41). Thus, the role of miR-31–5p/CTR axis and the miR-31–5p/CTR blocking strategies may be of relevance and interest to the (patho)physiology of skeletal system.
We also noted that a prominent reduction in atrial fibrosis was associated with a relatively modest decrease in AF burden in older mice. This might be due to the relatively short (3.5 weeks) TSB treatment period or the insufficient CTR activation by its ligand CT, which is progressively suppressed in Lkb1-aKD mice. Thus, it is possible that a combined therapeutic approach of miR-31/CTR-TSB and hormonal CT replenishment (with clinically approved CT-analogues) might exert an even more potent anti-fibrotic and anti-arrhythmic effect. The results presented here, taken in the context of the existing evidence demonstrate the specificity of the miR31/CALCR-TSB approach and position CALCR as an important miR-31–5p downstream target in the context of fibrosis and arrhythmia. Together these data suggest enrichment of this miR in the atrial myocardium compared to LV. Furthermore, given that miR-31–5p is highly enriched in the atrial myocardium, strategies to manipulate miR-31–5p can offer primarily atrial-targeted therapeutic options, allowing to circumvent the non-specific effects on the ventricular myocardium – a serious limitation of the current antiarrhythmic drugs.
In summary, we have identified a novel miR-31–5p/CTR-mRNA regulatory mechanism upstream of the cardiac calcitonin system in human atrial myocardium. Furthermore, we found that advanced atrial fibrosis, commonly seen in persAF, is characterised by hACFs enriched for miR-31–5p which, via binding to the CTR-3’UTR, activates fibroblast proliferation and collagen production. Selective disruption of miR-31–5p/CTR-mRNA binding reversed atrial fibrosis and decreased AF burden in vivo in a clinically relevant model of progressive atrial fibrosis and arrhythmia. Thus, selective blockade of the miR-31–5p/CTR-mRNA interaction with miR-31/CTR-TSB treatment, potentially in combination with currently approved hormonal CT-therapy, may offer a safer, atrial-selective strategy to restore CT signalling and re-establish control over atrial fibrogenesis and prevent associated arrhythmia susceptibility in patients with a history of AF. The miR31/CTR-TSB offers a target-selective approach in miR therapeutics, which will have to be further validated in human AF in clinical trials.
Supplementary Files
This is a list of supplementary files associated with this preprint. Click to download.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Ball J, Carrington MJ, Mc Murray JJ, Stewart S. Atrial fibrillation: profile and burden of an evolving epidemic in the 21st century. Int J Cardiol. 2013; 167(5),1807–24.23380698 10.1016/j.ijcard.2012.12.093 · doi ↗ · pubmed ↗
- 2Chugh SS, Havmoeller R, Narayanan K, Singh D, Rienstra M, Benjamin EJ, Gillum RF, Kim YH, Mc Anulty JH Jr, Zheng ZJ, Forouzanfar MH, Naghavi M, Mensah GA, Ezzati M, Murray CJ. Worldwide epidemiology of atrial fibrillation: a Global Burden of Disease 2010 Study. Circulation. 2014; 129(8):837–47.24345399 10.1161/CIRCULATIONAHA.113.005119 PMC 4151302 · doi ↗ · pubmed ↗
- 3Wijffels MC, Kirchhof CJ, Dorland R, Allessie MA. Atrial fibrillation begets atrial fibrillation. A study in awake chronically instrumented goats. Circulation. 1995; 92(7):1954–68.7671380 10.1161/01.cir.92.7.1954 · doi ↗ · pubmed ↗
- 4Nattel S. New ideas about atrial fibrillation 50 years on. Nature. 2002; 415(6868):219–26.11805846 10.1038/415219 a · doi ↗ · pubmed ↗
- 5Clarnette JA, Brooks AG, Mahajan R, Elliott AD, Twomey DJ, Pathak RK, Kumar S, Munawar DA, Young GD, Kalman JM, Lau DH, Sanders P. Outcomes of persistent and long-standing persistent atrial fibrillation ablation: a systematic review and meta-analysis. Europace. 2018; 20(FI_3):f 366–f 376.29267853 10.1093/europace/eux 297 · doi ↗ · pubmed ↗
- 6Chelu MG, King JB, Kholmovski EG, Ma J, Gal P, Marashly Q, Al Juaid MA, Kaur G, Silver MA, Johnson KA, Suksaranjit P, Wilson BD, Han FT, Elvan A, Marrouche NF. Atrial Fibrosis by Late Gadolinium Enhancement Magnetic Resonance Imaging and Catheter Ablation of Atrial Fibrillation: 5-Year Follow-Up Data. J Am Heart Assoc. 2018; 7(23):e 006313.30511895 10.1161/JAHA.117.006313 PMC 6405558 · doi ↗ · pubmed ↗
- 7Heijman J, Guichard JB, Dobrev D, Nattel S. Translational Challenges in Atrial Fibrillation. Circ Res. 2018; 122(5):752–773.29496798 10.1161/CIRCRESAHA.117.311081 · doi ↗ · pubmed ↗
- 8Nattel S. Molecular and Cellular Mechanisms of Atrial Fibrosis in Atrial Fibrillation. JACC Clin Electrophysiol. 2017; 3:425–435.29759598 10.1016/j.jacep.2017.03.002 · doi ↗ · pubmed ↗