The Establishment of Prostate-specific, SKP2 Humanized Mice by CRISPR Knock-in Method Reveals Neoplastic Initiation and Microenvironmental Reprogramming
Liankun Song, Yurong Song, Vyvyan Nguyen, Shan Xu, Kelly Ho, Altaf Mohammed, Robert H. Shoemaker, Bang H. Hoang, Jianhua Yu, Edward Uchio, Xiaolin Zi

TL;DR
Scientists created a mouse model with human SKP2 to study prostate cancer initiation and changes in the tumor environment.
Contribution
A prostate-specific human SKP2 knock-in mouse model was established, revealing neoplastic initiation and microenvironmental changes.
Findings
Overexpression of hSKP2 induces PIN and low-grade carcinoma in mice.
RNA-sequencing showed altered EMT, extracellular matrix, and interferon signaling genes.
SKP2 inhibitors Flavokawain A and C1 reduced viability of hSKP2 organoids.
Abstract
Genetic inactivation of SKP2 has been shown to effectively prevent cancer initiation and block tumorigenesis. However, direct in vivo evidence for SKP2 on cancer initiation and prostatic microenvironment is still lacking and a SKP2 humanized mouse model is critical for developing prostate cancer immunoprevention approaches through targeting SKP2. We therefore have established a prostate-specific human SKP2 knock-in mouse model driven by an endogenous mouse probasin promoter. Overexpression of hSKP2 induces PIN and low-grade carcinoma. RNA-sequencing analysis revealed significant gene expression alterations in EMT, extracellular matrix, and interferon signaling. Single cell deconvolution showed an increase of fibroblast population and a decrease of CD8+ T cell and B cell populations. Consistently with these results from the SKP2 humanized mouse, SKP2 protein is overexpressed in human…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
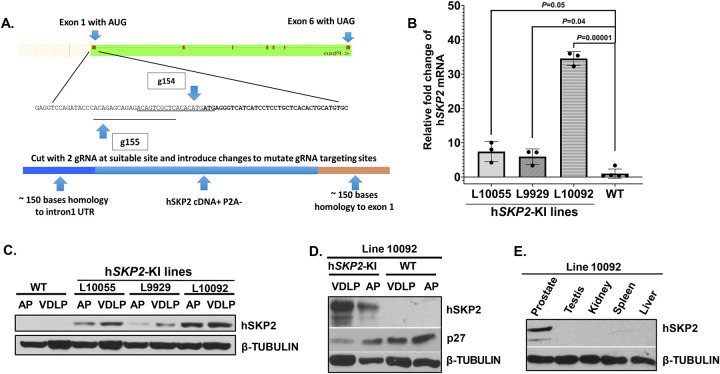 Figure 1
Figure 1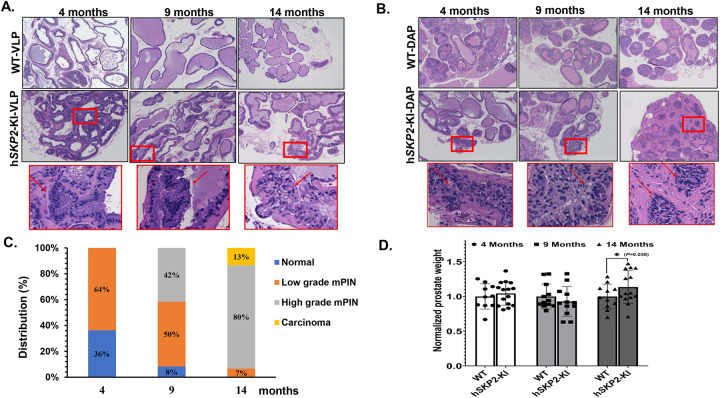 Figure 2
Figure 2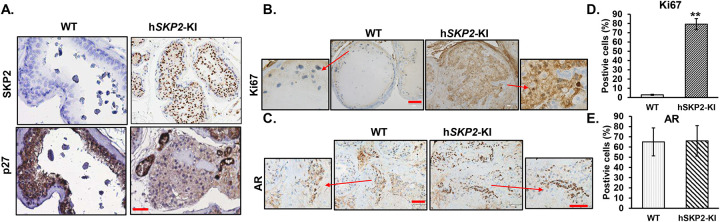 Figure 3
Figure 3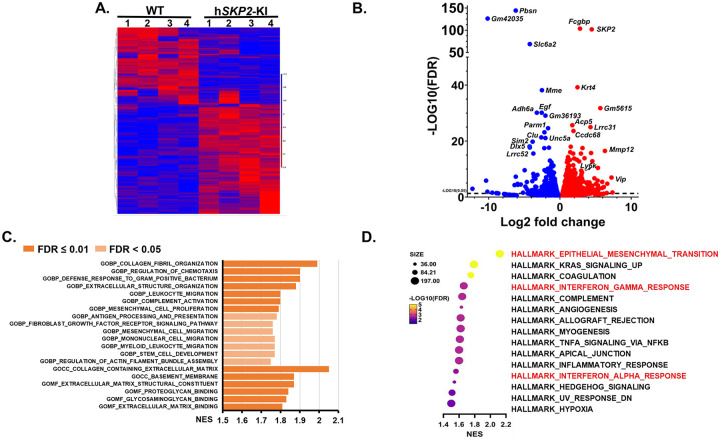 Figure 4
Figure 4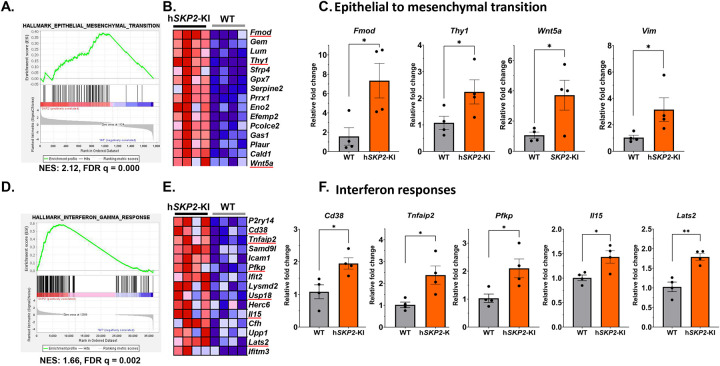 Figure 5
Figure 5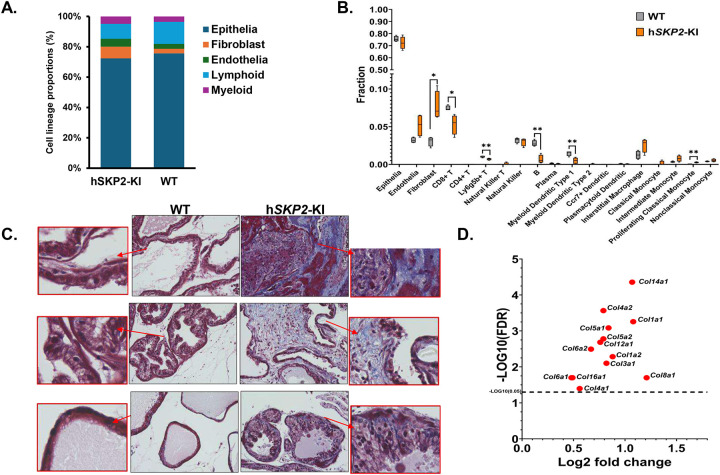 Figure 6
Figure 6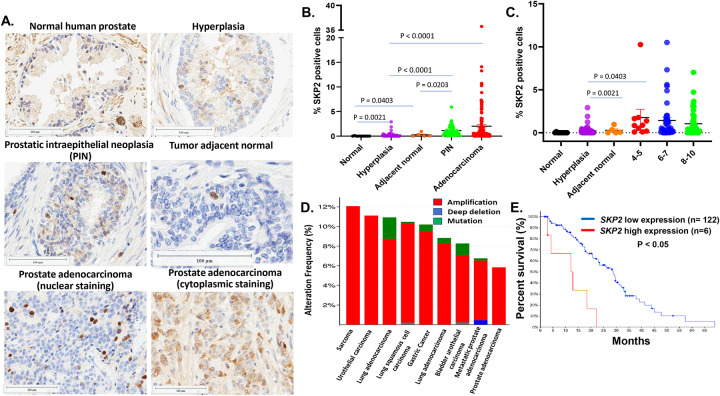 Figure 7
Figure 7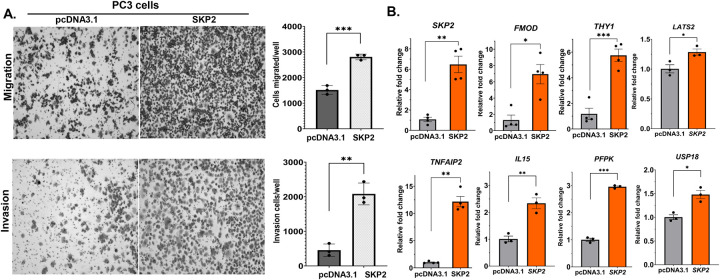 Figure 8
Figure 8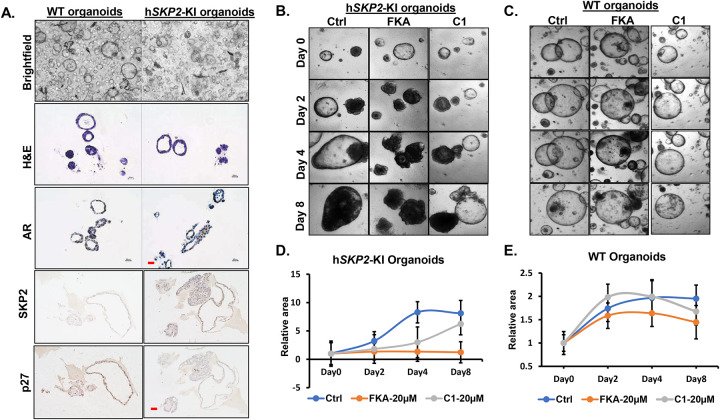 Figure 9
Figure 9Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsProstate Cancer Treatment and Research · Connective Tissue Growth Factor Research · Wnt/β-catenin signaling in development and cancer
Introduction
S-phase kinase associated protein 2 (SKP2) is the rate-limiting component of the Skp, Cullin, and F-box (SCF) containing E3 ubiquitin ligase complex that catalyzes the ubiquitination of proteins for targeted degradation by 26S proteasome [1–3]. SKP2 plays a central role in cell cycle regulation, cellular senescence, and apoptosis by targeting and ubiquitinating cell-cycle and apoptosis regulators, including E2F1, the Forkhead box O (FOXO) family members. SKP2 binds and degrades cyclin-dependent kinase inhibitors such as p27^Kip1^ and p21 for controlling cell cycle entry and G1/S transition and then cell proliferation [1–3].
SKP2 has been shown to function as a proto-oncogene causing the pathogenesis of lymphomas [4]. Overexpression of SKP2 is frequently observed and associated with cancer progression, metastasis, and poor prognosis in a wide variety of cancers including prostate cancer [4, 5]. In clinical studies of prostate cancer, SKP2 has been reported to be a key amplified gene [6]. SKP2 overexpression has been found in 86.4 % (64 of 74 samples) of pre-malignant high grade-prostatic intraepithelial neoplasia (HG-PIN) and in 557 of 622 (89.5%) primary prostate cancer specimens, showing a strong positive association with pre-operative serum prostate specific antigen (PSA) levels, Gleason score, tumor grade, and biochemical failure in men treated by prostatectomy [7–11]. There are numerous studies using a Skp2 knockout mouse model. These studies demonstrate that Skp2 deficiency in mouse models abolishes or inhibits spontaneous tumorigenesis that were initiated by inactivation of PTEN, ARF, pRB as well as by overexpression of Her2, through inducing p53-independent cellular senescence or blocking Akt-mediated aerobic glycolysis [12–15]. A very recent study published in Nature by Chen et al. [16] reported that cells with shorter cell cycle durations are more susceptible to oncogenic transformation than those with longer cell cycles and genetic inactivation of Skp2, a key determinant of cell cycle duration, completely block tumorigenesis in mice. These results suggest a previously unrecognized mechanism for developing new cancer preventive or therapeutic strategies through targeting SKP2.
However, compared to large amounts of studies using a Skp2 knockout mouse model, direct in vivo evidence for human SKP2 on tumor initiation and prostatic microenvironment is still lacking. In addition, to develop prostate cancer immunoprevention and immunotherapy approaches by targeting human SKP2 oncogene and its associate pathways, a prostate-specific SKP2 humanized mouse model is critical. We therefore have established a more pathophysiologically relevant prostate carcinogenesis model by knocking-in of human SKP2 gene into a locus that is regulated by the endogenous probasin promoter using the CRISPR/Cas9 method. We have also characterized molecular and cell population alterations in the prostate of human SKP2 (hSKP2-KI) mice. The results demonstrate a profound remodeling of the prostatic microenvironment through hSKP2 overexpression-induced dysregulation of EMT and interferon pathways. Single cell deconvolution shows an increase of fibroblast population and a decrease of CD8^+^ T cell and B cell populations in the prostates of hSKP2-KI mice. Meanwhile, SKP2 overexpressing prostate organoids were developed for convenient and efficient screening of selective SKP2 inhibitors, which were validated by testing Flavokawain A (FKA), a naturally occurring chalcone in the Kava plant that acts as a SKP2 degrader, as we previously reported [17]. Consistent with our previous findings, FKA significantly inhibits growth of SKP2 overexpressing prostatic organoids, while having minimum toxicity on the normal prostate organoids. Our data suggest that the humanized hSKP2-KI mouse model and the organoids derived from the model are both valuable tools to study prostate cancer and test prostate cancer prevention and interception agents.
Materials and Methods
Establishment of prostate-specific human SKP2 knock-in mouse model
P2A-Human SKP2 coding sequence was knocked into the endogenous mouse probasin at exon 1 locus by a CRISPR/Cas9-based approach by the University of California Irvine Transgenic Mouse Facility. After sequencing, heterozygous hSKP2-KI females were crossed with wild-type FVB/N male mice to generate male offspring specifically expressing hSKP2 in all lobes of mouse prostates. Mice were identified with ear tagging and the tail DNA was subjected to polymerase chain reaction (PCR) with the following primers: primer 845 (5’-ATACCTGAAACATGGGATAGGCAC-3’), 846 (5’-TTGCTACTCAGGTCTGGAATCTC-3’) and 847 (5’- ACCAAGTTCCAGAACATTCGTTTC-3’). Prostate lobes of hSKP2-KI and wild type (WT) male mice at different ages (4, 9 and 14 months) were dissected, weighed and fixed with 10% neutral buffered formalin for histological and immunohistochemistry analyses or snap frozen for RNA and protein extraction. Prostate-specific expression of human SKP2 was confirmed by quantitative PCR, Western blotting, and immunohistochemical staining. Histological evaluation of mouse prostatic lesions was performed according to the publication by Dr. Cardiff [18]. Animal care and experiments were carried out according to institutional guidelines and the approved protocol by University of California, Irvine (protocol #: AUP-19–109).
Quantitative PCR assay
PC3 cell line (RRID:CVCL_0035) was purchased from American Type Culture Collection (ATCC, Manassas, VA) and stably transfected with PcDNA3.1 vector control and SKP2. These cell lines were tested for known species of mycoplasma contamination using a kit from LONZA Inc. (Walkersville, MD) every six months. The cell line used was mycoplasma contamination free. Total RNAs were extracted immediately from the dissected prostate lobes of WT and hSKP2-KI mice and human prostate cancer cell lines of PC3/PcDNA3.1 and PC3/SKP2 using RNAStat (Amsbio, Friendswood, TX). The primer sequences were designed using PrimerBank (https://pga.mgh.harvard.edu/primerbank/) and the specificity of the primers was checked using nucleotide BLAST in the NCBI. The primer sequences were listed in supplementary table 1. Primers used for the quantitative PCR were purchased from OriGene (Rockville, MD) or synthesized by Eurofins Genomics (Louisville, KY). Oligo-dT GoScript^™^ Reverse Transcription mix (Promega, Mandison, WI) was used for the synthesis of the first-strand cDNA. The iTaq SYBR green super-mix (BioRad, Hercules, CA) was used in the real-time PCR reaction on CFX Connect Real-Time System (BioRad, Hercules, CA). Relative quantitative fold changes compared to control were calculated using the comparative 2^−ΔΔCt^ method [19]. Mouse Gapdh and human *ACTB (*β-ACTIN) gene were used as internal controls.
Western blot analysis
Prostate tissues from the prostate lobes of WT and hSKP2-KI mice were homogenized in radioimmunoprecipitation assay (RIPA) buffer with protease inhibitors, and protein concentrations were determined by Bio-Rad DC protein assay (BioRad, Hercules, CA). Equal amount of total protein was loaded onto denaturing SDS-polyacrylamide gel (8–16%) for electrophoresis and then transferred onto PVDF membranes as described previously [20]. After blocking with 5% non-fat milk, the membranes were probed with antibodies against SKP2 (Abcam Inc, Waltham, MA), p27^Kip1^ (BD Biosciences, Franklin Lakes, NJ), androgen receptor (AR) (Cell Signaling Technology, Beverly, MA), or β-TUBULIN (Santa Cruz Biotechnology, Dallas, TX) overnight at 4°C. The membranes were then incubated with corresponding anti-mouse or anti-rabbit secondary antibodies (Cell Signaling Technology, Beverly, MA) followed by wash and visualization by Prometheus Protein ProSignal^®^ Dura ECL Reagent (Genesee Scientific, El Cajon, CA) on X-ray films.
Immunohistochemistry (IHC) analysis
Prostate tissues were fixed in 10% neutral buffered formalin, paraffin embedded and sectioned at 5μm thickness. Sections were deparaffinized, hydrated, followed by antigen retrieval with sodium citrate buffer for 20 minutes in a steamer. The sections were then quenched with 3% hydrogen peroxide for 10 min followed by three washes of PBS and blocked with 3% normal goat serum for one hour. Afterwards, the slides were incubated with primary antibodies against Ki67 and SKP2 (Cell Signaling Technology, Danvers, MA) and p27^Kip1^ (BD Biosciences, Franklin Lakes, NJ) and with horseradish peroxidase (HRP) labeled secondary antibody. Sections were visualized with 3, 3-diaminobenzidine (DAB) using the Cell and Tissue Staining kit (R&D Systems, Minneapolis, MN) and imaged under Keyence BZ-X710 microscope (Itasca, IL). The images are quantified in Image J by counting the total number of positively stained cells at 12 arbitrarily selected fields at ×40 magnification in a double-blind manner. Images from 3 mice per group were used for statistical analysis. In addition, human prostate tissue microarrays (TMA) were purchased from Tissue Array LLC (TissueArray.com, Derwood, MD) with different pathology including hyperplasia, PIN, adenocarcinoma, and normal prostate tissues. SKP2 IHC was performed using a Leica Bond RX autostainer (Leica Biosystems, Deer Park, IL). Antigen retrieval was performed using EDTA for 20 minutes at 100°C on the Bond autostainer. Slides were removed from the autostainer for primary SKP2 antibody (Cell Signaling # 2652; 1:100 dilution) incubation overnight at 4°C. Slides were reloaded on the Bond autostainer. Antibody detection was then accomplished using the Bond Polymer Refine Detection kit (Leica Biosystems #DS9800) with the post-primary reagent removed from Leica’s default staining protocol. Slides were scanned, then analyzed and quantified using HALO software.
RNA sequencing and analysis
Ventral lobes of prostates from hSKP2-KI (n=4) and WT mice (n=4) at 12 months old age were freshly dissected and immediately homogenized in RNAStat (Amsbio, Friendswood, TX). The integrities of total RNA were analyzed by Agilent 2100 with the RNA integrity numbers ranging from 8 to 10. mRNA was purified using poly-T oligo-attached magnetic beads and after fragmentation, the first strand cDNA was synthesized using random hexamer primers followed by the second strand cDNA synthesis. The library was ready after end repair, A-tailing, adapter ligation, size selection, amplification, and purification. Libraries were then checked with Qubit and real-time PCR for quantification and bioanalyzer for size distribution, then they were pooled and sequenced on Illumina Platform PE150 (20 million paired-end reads) by Novogene Corporation Inc (Sacramento, CA). The raw paired-end reads were trimmed and quality checked in CLC Genomic Benchwork (Qiagen, Hilden, Germany), followed by mapping to the mouse genome. Normalized counts were used to analyze the gene expression patterns. The raw and normalized data files are deposited in Gene Expression Omnibus (GEO) with series entry number GSE295398. Differently expressed genes (DEGs) in hSKP2-KI mouse prostate samples were identified compared to WT mouse prostate samples with the false discovery rate (FDR) <= 0.05. Gene ontology (GO) and hallmark pathways were analyzed using Gene Set Enrichment Analysis software (GSEA) from the Broad Institute. Enriched pathways were treated as significant with FDR <= 0.05.
Migration and invasion assay
Falcon^®^ HTS 24-well Multiwell system (Cat# 351185, Corning, AZ) and Corning^®^ BioCoat^™^ Matrigel^®^ Invasion Chamber (Cat#354480, Corning, AZ) were used for migration assay and invasion assay, respectively, as described previously [21]. After cell starvation for 24 hours, 30,000 PC3/pcDNA3.1 and PC3/SKP2 cells were seeded onto the upper insert in 500μl RPMI1640 medium without serum, and 750 μl RPMI 1640 with 10% FBS was added to the bottom well as chemoattractant or RPMI 1640 without serum as control well. After 72 hours, non-migrating or non-invading cells were removed from the upper surface of the membrane by moistened cotton swab scrubbing followed by cell fixation with cold methanol and staining with hematoxylin. Images from 5 different areas of each insert were acquired and cell numbers of 3 replicate inserts from each cell line were used for statistical analysis.
Culture of prostate organoids and evaluation of SKP2 inhibitors’ anti-tumor effects
We have followed the protocol that was published by Hans Clevers’ group [22]. Fresh prostate tissues from SKP2-KI and WT male mice were minced into small pieces and digested in 5 mg/ml Collagenase II (Life Technologies, Carlsbad, CA) solution containing 10μM Y-27632 (AbMole BioScience, Houston, TX) for 2 hours at 37°C with shaking. After washing with adDMEM/F12+/+/+ medium containing penicillin/streptomycin, 10mM HEPES, and 1X GlutaMAX (Life Technologies, Carlsbad, CA), the pellets were further digested in TrypLE solution (Life Technologies, Carlsbad, CA) containing 10μM Y-27632 for approximately 15 minutes at 37°C with pipetting every 5 minutes, followed by washing and centrifugation. 20,000 digested cells were resuspended in cold Matrigel and plated 20μl/well into prewarmed 48 well plates. The plates were then upside down in the 37°C incubator for 15 min to allow the Matrigel to solidify. 300μl-500μl pre-warmed mouse prostate culture medium plus 10μM Y-27632 was added into each well. Culture medium was refreshed every 2–3 days. TrypLE solution supplemented with Y27632 was used for passaging the organoid culture. WT and hSKP2-KI prostate organoids were fixed with 4% paraformaldehyde, embedded in HistoGel (Thermo Scientific, MA) and sectioned for H&E staining for histological evaluation and for IHC analysis of AR, SKP2 and p27^Kip1^ expression.
To evaluate the effects of known SKP2 inhibitors FKA and C1 on the growth of prostate organoids, hSKP2-KI prostate organoids and WT prostate organoids were plated into 96 well plates and then treated with vehicle control (0.5% DMSO) or FKA or C1 inhibitor at indicated concentrations in triplicate. The morphology of the organoids was imaged before the treatments and at different time-points after the treatments under Keyence fluorescence microscopy. The size of each organoid was analyzed by Image J software. In addition, the viability of the organoids was determined by measuring fluorescence intensities from the conversion of a cell-permeant nonfluorescent Calcein AM dye (CalAM, Thermo Scientific, MA) into a green fluorescent Calcein (ex/em 495 nm/515 nm) in living cells. The fluorescence images were obtained at different treatment times under Keyence fluorescence microscopy and analyzed by Image J. Fluorescence intensity and size of each organoid acquired at different time points were compared to its intensity and size at day 0 to obtain the relative changes over time (n=15 for WT or KI prostate organoids).
Cell deconvolution of bulk RNA-seq data
Mouse prostate single cell signature was obtained from the study by Joseph DB et al [23]. The mouse prostate single cell annotation was downloaded from GUDMAP database at https://doi.org/10.25548/17-DRBC, The rds file was processed in R software and mouse prostate single cell genes and cells annotation matrix were extracted as the signature file used in CIBERSORTx (https://cibersortx.stanford.edu/). Bulk RNA-seq data from WT and hSKP2-KI mouse prostates were used as a mixture input according to the CIBERSORTx tutorial. S-mode batch correction was applied and estimation of cell fractions from bulk tissue transcriptomes was generated on CIBERSORTx as described in published paper [24].
Statistical analysis.
Student’s t-test, one-Way ANOVA followed by the Bonferroni t-test for multiple comparisons and log-rank testing for survival analysis were carried out using GraphPad Prism 9.0 to calculate statistical significance. Data are expressed as mean ± standard deviation (SD). All statistical measures were two-sided, and p-values <0.05 were statistically significant.
Results
Establishment of prostate specific human SKP2-KI mouse lines by a CRISPR/Cas9 method
The P2A-hSKP2 coding sequence was introduced into the guide RNA generated DNA break by a CRISPR/Cas9 method in the endogenous mouse probasin at exon 1 locus (Fig. 1A) for expression of hSKP2 under control of the mouse probasin promoter. Probasin-hSKP2-KI mouse lines were screened using specific PCR of genomic DNA extracted from mouse tail biopsy and confirmed the correct sequencing of the genomic insert by the Sanger sequencing. Nineteen of 43 (44.2%) KI founder mice were genotyped positive. Three probasin-hSKP2-KI mouse lines (L9929, L10055 and L10092) show 5, 7-to-35-fold overexpression of hSKP2 in the prostate compared to that in the prostate of wild-type mice (Fig.1B). Western blotting analysis using human specific anti-SKP2 antibody (Abcam #ab68455) reveals that human SKP2 protein is expressed in the microdissected prostate lobes (anterior, dorsal, lateral, and ventral lobes) of three probasin-hSKP2-KI mouse lines but not in the prostate of wild-type mice (Fig. 1C). The expression level of hSKP2 protein is lower in the anterior lobe compared to other prostate lobes (Fig. 1C). Consistently, p27^Kip1^ protein, a putative substrate of SKP2 is down-regulated in the prostate of probasin-hSKP2-KI line L10092 (Fig. 1D). There is no detectable hSKP2 protein in other organs of the L10092 line, including testis, kidney, spleen, and liver (Fig. 1E). These results indicate that SKP2 overexpression is prostate-specific in the probasin-hSKP2-KI lines.
SKP2 overexpression induces hyperplasia, mPIN and low-grade carcinoma associated with increased SKP2 expression and cell proliferation and decreased expression of p27Kip1.
Histological evaluation shows that H&E-stained prostate lobes (anterior, dorsal, lateral, and ventral lobes) of probasin-hSKP2-KI mice at different ages (4, 9, and 14 months of age) exhibit different grades and proportions of marked enlargement and variation in the duct size, thicker and disoriented epithelial layers, primitive cribriform patterns, filled duct lumens, vacuolated cytoplasm, prominent round to oval nuclei with one or more nucleoli, and multiple mitotic figures, while similar lesions were not found in the prostate of wild type mice at the same ages (Figs. 2A & B). At 4 months of age, 63.3 % (7 out of 11) of probasin-hSKP2-KI mice were observed with hyperplasia and low-grade mPIN; 50% (6/12), 42% (5/12), and 8% (1/12) of probasin-hSKP2-KI mice with low-grade PIN, high-grade PIN, and hyperplasia, respectively, at 9 months of age; 6.7% (1/15), 80% (12/15) and 13.3% (2/15) of probasin-hSKP2-KI mice with low grade PIN, high grade PIN and low-grade carcinoma at 14 months of age, respectively (Fig. 2C). The mean prostate weights of probasin-hSKP2-KI mice were significantly increased compared to those of wild-type mice (P < 0.05) at 14 months of age (Fig. 2D).
IHC analysis reveals that strong SKP2 positive staining was mainly observed at the sites of prostatic lesions (i.e. hyperplasia, PINs and low-grade carcinoma), accompanied by decreased staining intensities of p27^Kip1^ in probasin-hSKP2-KI mice compared to those from wild-type mice (Fig. 3A). In addition, cell proliferation was significantly enhanced in the prostates of probasin-hSKP2-KI mice as indicated by increased Ki67 positive staining cells by approximately 26 folds compared to those from wild-type mice (P < 0.01) (Figs 3B & D), while there was no significant difference in AR expression in the prostates between probasin-hSKP2-KI mice and wild-type mice (Figs. 3C & E).
Gene expression profiling reveals differentially expressed genes and significant enrichment of EMT and interferon pathways in SKP2 overexpressing mouse prostates
We performed bulk RNA sequencing to determine the effect of SKP2 overexpression on global gene expression alterations in the prostate of probasin-hSKP2-KI mice and wild-type mice. A total of 1,753 differentially expressed genes (DEGs) were identified with a false discovery rate (FDR) p-value < 0.05. Among them, 678 genes demonstrated more than a two-fold change in expression, including 498 genes being up-regulated and 180 genes downregulated (Figs. 4A & B). The top DEGs include Fcgbp, Krt4, Slc6a2, Mme, Egf, Adh6a, Clu, Lrrc31, Sim2, Mmp12, Ly6k, Ly6d, Ly6a, etc. Androgen responsive genes (Ccnd1, Hmgcr, Tmprss2, Insig1, Dhcr24, and Bmpr1b), G2M related genes (Slc7a1, Egf, and Cul4a) and apoptosis related genes (Clu, Bcl2, and Sc5d) are also on the top list of DEGs. The Gene Ontology (GO) gene set enrichment analysis (GSEA) highlights the activation of extracellular matrix components, leukocytes migration, and mesenchymal cell proliferation pathways in SKP2 overexpressing mouse prostates (Fig. 4C). In addition, GSEA of hallmark genes revealed that EMT, interferon alpha and gamma, angiogenesis, and inflammatory response pathways pathway are significantly enriched in SKP2 overexpressing mouse prostates (Fig. 4D).
The list of enriched gene expression alterations in EMT and interferon pathways is shown as enrichment profiles and heatmaps in Figs. 5A, B, D and E. The increased gene expression levels of selected EMT pathway genes, including Fmod, Thy1, Wnt5a, and Vim, and interferon gamma pathway genes, such as Tnfaip2, Cd38, Pfkp, Il15, Lats2 were verified by quantitative PCR method (Figs. 5C & F and supplementary Fig. 1).
Cell deconvolution analysis reveals an increase in fibroblasts and a decrease in infiltrating T and B cells in SKP2-overexpressing mouse prostates
Given the profound alterations in gene expression of EMT, extracellular matrix and interferon pathways in SKP2 overexpressing mouse prostates, we have deconvoluted the probasin-hSKP2-KI and WT bulk RNA-seq data using the R package CIBERSORTx. The cell deconvolution analysis revealed significant differences in the proportional compositions of various immune and stromal cell types in the prostate of the hSKP2-KI versus WT mice (Fig. 6A). The hSKP2-KI mice exhibit a significantly higher proportion of fibroblasts in the prostate compared to that of WT mice (P = 0.0122, Student’s t test) (Fig. 6B). A reduction in lymphoid lineage cells and an expansion of myeloid lineage cells have also been observed in the prostate of hSKP2-KI mice compared to that of WT mice. Notably, hSKP2-KI mouse prostate glands demonstrate a significantly lower proportion of CD8^+^ T cells, B cells, and myeloid dendritic type I cells compared to WT mouse prostate glands (Ps = 0.0435 to 0.0012, Student’s t test). Proliferating classical monocytes that have the potential to differentiate to tumor-associated macrophages, which suppress T cell function, [25] were found to infiltrate into the prostate of hSKP2-KI mice more than that of WT mice (P = 0.008, Student’s t test).
We have further evaluated the extracellular (i.e. collagen) components of hSKP2-KI and WT mouse prostates using Masson’s Trichrome staining and expression of collagen related genes. Figures 6C and 6D show a markedly enhanced collagen staining and upregulation of numerous collagen related genes in the prostate of hSKP2-KI mice versus WT mice. Taken together, these results suggest that SKP2 overexpress induces the remodeling of ECM and creates an immune suppressive prostatic microenvironment to support tumor initiation and development.
SKP2 is sparsely overexpressed in human prostate hyperplasia, PIN and prostate adenocarcinoma compared to prostate normal tissues; Higher levels of SKP2 mRNA in prostate tumor tissues are associated with poorer survival of prostate cancer patients.
Next, we examined the expression status of SKP2 on TMAs of prostate tissues with different pathology including hyperplasia, tumor adjacent tissue, PIN, adenocarcinoma, and normal prostate tissues. SKP2 is sparsely overexpressed in hyperplasia, tumor adjacent tissues, PIN and adenocarcinoma with average percentage of positive SKP2 staining cells per core of 0.34%, 0.46%, 1.16%, and 2.43%, respectively, compared to that of normal prostate tissues (0.12%) (Fig. 7A & B) (Ps < 0.05). 10% (1/10) of prostate normal tissue cores, 68.6% (33/49) of hyperplasia, 60% (3/5) of tumor adjacent tissues, 97.6% (41/42) of PIN, and 81% (113/138) prostate adenocarcinoma tissue cores were positive for SKP2 (Table 1). There is no significant difference in SKP2 expression among prostate adenocarcinoma tissue cores with different Gleason scores (Fig. 7C). The vast majority of positive SKP2 staining are localized in nucleus and a few cores of prostate adenocarcinoma exhibit cytoplasmic staining (Fig. 7A). The staining patterns of SKP2 in different prostate pathology in humans are consistent with the results from the probasin-SKP2-KI mice (Figure 3A). Our results suggest SKP2 overexpression is an early event in prostate carcinogenesis.
In addition, we explored the cBioport dataset for the frequencies of SKP2 genetic alterations (i.e. amplification, deep deletion, and mutation) in cancers. We observed that amplification of SKP2 frequently occurs in various cancers, including sarcoma, urothelial or bladder cancer, lung cancer, gastric cancer, metastatic prostate cancer, and prostate adenocarcinoma with a range from about 12% to 6% (Fig. 7D). Analysis of RNA expression levels with available survival data in the SU2C/PCF Dream Team, PNAS 2019 study shows that higher levels of mRNA levels in prostate tumor tissues are significantly associated with poorer survival of prostate cancer patients (P < 0.05, Log-rank test) (Fig 7E). These results suggest that SKP2 overexpression is an unfavorable prognostic factor associated with disease aggressiveness.
SKP2 overexpression promotes cell migration and invasion, and regulates the expression of EMT, and genes related to interferon signaling.
To further evaluate the association of SKP2 overexpression with aggressiveness of prostate cancer, we have performed cell migration and invasion experiments, as well as quantitative PCR analysis of the expression of EMT and interferon pathways related genes using a prostate cancer cell line PC3 with or without SKP2 overexpression. Figure 8A shows that SKP2 overexpression increases cell migration and invasion of PC3 cells by 1.8 and 4.5 folds, respectively, compared to PC3 cells expression vector control pcDNA3.1 (Ps < 0.01, Student’s t test). In addition, SKP2 overexpression in PC3 cells increases the expression of EMT and interferon gamma pathway related genes as described above in the hSKP2-KI mouse model, including FMOD, THY1, TNFAIP, IL15, PFKP, USP18, and LATS2 (Fig. 8B). Taken together, data from the hSKP2-KI mouse model, human prostate cancer clinical data and in vitro human prostate cancer cell culture studies are consistent, at least in part, to support the critical role of SKP2 overexpression promote aggressiveness of prostate cancer by regulating the EMT and interferon pathways. Our data suggest that the humanized mouse model recapitulates human prostate cancer.
Generation and characterization of prostate organoids derived from hSKP2-KI mice for testing SKP2-targeting agents
Since in vivo studies of SKP2 targeting agents using mouse models are often time consuming and cost-prohibitive, we therefore established cultures of prostate organoids from the prostate of both hSKP2-KI and WT mice to facilitate screening of selective SKP2 targeting agents. H & E staining and histological analysis shows that the morphology of prostate organoids from WT mice is characterized by a branched, acinar-like structure and mimics the in vivo the epithelial architecture of mouse prostate tissue, while prostate organoids from hSKP2-KI mice display a morphology of a distorted acinar-like structure with multiple layers of cells and filled lumens (Fig. 9A). Prostate organoids from hSKP2-KI mice also show strong expression of SKP2 accompanied by negative staining of p27^Kip1^, whereas prostate organoids from WT mice exhibit the opposite expression status of SKP2 and p27^Kip1^. AR expression is also positive in prostate organoids derived from both SKP2-KI and WT mice (Fig. 9A).
We have previously reported that FKA acts as a SKP2 degrader for selectively inhibiting the growth of prostate cancer PC3 cells overexpressing SKP2 [17]. In addition, SKP2 inhibitor C1 has been shown to specifically target the interaction between SKP2 and p27^Kip1^, preventing the binding and subsequent ubiquitination and degradation of p27^Kip1^ [26]. To validate our organoid model, we therefore tested the selectivity of these known SKP2 inhibitors on the growth of SKP2 overexpressing prostate organoids versus normal prostate organoids to establish a “proof of concept” tool for future screening with more SKP2 inhibitors. Figures 9 B & C show that control-treated hSKP2-KI organoids continuously increase their sizes until day 4 after seeding, whereas FKA- and C1-treated hSKP2-KI organoids exhibit a significantly slower growth in size (Ps <0.05). However, C1-treated hSKP2-KI organoids recovered from their growth inhibition on day 8, whereas the mean sized of FKA treated hSKP2-KI organoids continued to decrease (Figures 9 D&E). This result suggests that hSKP2-KI organoids could potentially develop resistance to C1. In addition, both C1 and FKA exhibit no significant growth inhibitory effects on wild-type prostate organoids. FKA at 12.5 μM completely reduced cell viabilities of hSKP2-KI organoids 3 days after the treatment but had no effect on the growth of wild-type organoids (Supplementary Figure. 3). FKA exhibited excellent selectivity to the growth of hSKP2-KI organoids over wild-type organoids.
Discussion
Our analysis of publicly available datasets shows that SKP2 gene is frequently amplified in multiple cancers, including sarcoma, urothelial cancer, and prostate cancer, and that higher SKP2 mRNA levels in prostate tumor tissues are associated with poorer overall survival of prostate cancer patients. IHC analysis reveals that SKP2 is sparsely overexpressed in prostate hyperplasia, tumor adjacent tissues, PIN and prostate adenocarcinoma compared to normal prostate tissues. The densities of SKP2 positive cells progressively increase from normal prostate tissues to prostate hyperplasia, tumor adjacent tissues, PIN and prostate adenocarcinoma. Also, the percentages of prostate hyperplasia, tumor adjacent tissues, PIN and prostate adenocarcinoma tissues that have SKP2 positive cells ranging from 68.6% to 97.6% are much higher than normal prostate tissues (10%). In multiple studies, genetic inactivation of SKP2 can prevent tumor initiation and block tumorigenesis [14–16]. SKP2 affects the maintenance and expansion of stem cell pools and regulates pluripotency [27], which may explain its role in cancer initiation. We have also shown that genetic and pharmacological depletion of SKP2 reduces tumor-initiating properties and cancer stemness [28, 29]. Taken together, these results strongly support that SKP2 plays a key role in cancer initiation and can serve as an important target for cancer chemoprevention.
In addition, SKP2 interacts with pRB, PTEN, AR, and H-RAS in prostate cancer [4, 15, 30, 31]. Allelic loss or reduced expression of RB occurs in 25% to 50% of human prostate cancer [32–34], and PTEN deletion and/or mutations were detected in 30% of primary prostate cancer and 63% of metastatic prostate cancer [35, 36]. Genetic depletion or pharmacologic inhibition of SKP2 can block the pRB deficient tumorigenesis by inducing apoptosis or suppress cancer progression by triggering p53-independent senescence in prostates of Arf^−/−^ mice and PTEN deficient mice [2, 12, 14, 15, 37]. All evidence suggests that SKP2 functions as a key oncogene during prostate carcinogenesis and progression. SKP2 is also associated with the acquired drug resistance like paclitaxel resistance in prostate cancer [38, 39]. The acquired chemoresistance will eventually lead to treatment failure of castration-resistant prostate cancer [37, 38]. Since Skp2-knockout mice are viable and fertile [40], SKP2 may serve as an “Achilles’ heel” type of drug target to effectively prevent or treat pRB, p53 and/or PTEN deficient prostate cancer potentially with less toxicity. Therefore, there is an unmet need to develop a hSKP2-KI mouse model for further understanding the oncogenic functions of SKP2 in prostate carcinogenesis and to provide a novel tool for developing novel SKP2 inhibitors.
Previous prostate-specific transgenic mouse studies, such as TRAMP and Hi-Myc models, have used random integration of rat Pbsn mini promoter driven oncogenes into mouse genome, which requires considerable time, effort, and funds to screen many independent lines with a lower positive rate. In this study, we have developed the novel CRISPR/Cas9-based approach to knock-in a hSKP2 coding sequence into mouse Pbsn locus, such that hSKP2 expresses from the modified Pbsn locus. To the best of our knowledge, we are the first to use endogenous Pbsn to drive an oncogene expression specifically in the prostate of mice. Because Pbsn gene is X-linked, a solution to maintain transgenic line is to crossbreed of heterozygous females with wild type males. Since the CRISPR/Cas9 approach more accurately targets of hSKP2 to endogenous mouse Pbsn locus, the time and effort for screening of founder animals is significantly reduced compared to random insertion approaches. Indeed, our approach results in a significantly high rate (about 44.2%) of positive transgenic lines of hSKP2-KI and is more efficient compared to traditional transgenic approaches. In addition, endogenous mouse Pbsn promoter driven hSKP2 overexpression would be more physiopathologically relevant and provide more reproducible phenotypes and molecular signatures compared to traditional random insertion approaches into an unknown locus(s). In our study, hSKP2 is specifically overexpressed in all lobes of mouse prostate, but not in other organs, such as testis, kidney, spleen, and liver. SKP2 overexpression in mouse prostates induces atypical cell proliferation, leading to a progressive development of spontaneous prostatic lesions including hyperplasia, mPIN, and low-grade prostatic adenocarcinoma over 14 months of age. Compared to the ARR2PB (the rat probasin) promoter driven hSKP2 transgenic mice [41], our probasin-hSKP2-KI mice similarly develop hyperplasia, low grade PIN, high grade PIN and low-grade carcinoma, but exhibit enlarged prostate glands and low-grade carcinoma in a slower fashion at the advanced age of 14 months. The downregulation of p27^Kip1^, a key substrate of SKP2 was also observed to be restricted in the hyperplastic and dysplastic regions, accompanied by SKP2 overexpression and a marked increase in Ki67 positively staining cells.
The previously unreported discovery in this study stems from the RNAseq analysis, which have identified the top enrich pathways, including EMT, collagen fibril organization, interferon α/γ pathways, in prostate tissues from the hSKP2 KI vs. wild-type mice. Deconvolution of bulk RNA seq data into single cell distribution shows a significant increase in fibroblast population and proliferative classic monocytes and a decrease in CD8^+^ T cells, B cells, and myeloid dendritic type I cells in the prostate of the hSKP2 KI mouse. Ruan et al. [42] reported that SKP2 stabilized the TWIST1 protein, a master regulator of EMT, through a non-degradative ubiquitination to promote EMT and acquisition of cancer stem cell properties. We also show that SKP2 overexpression in prostate cancer PC3 cells increases the protein levels of TWIST1 and the expression of EMT related genes, including FMOD, THY1, NFIL3 and IRF2, leading to a marked increase in migration and invasion. You et al. reported that TWIST1 regulated the basal expression levels of interferon regulatory factor 9 (IRF9) and interacted with IRF9 to promote IRF9 binding to the IFN-stimulated response element [43]. Other studies also showed that SKP2 interacts with IFN-I receptor 2, T-box expressed in T cells, and SH2-containing protein tyrosine phosphatase-2 to regulate IFN-a/γ abundance [44, 45]. This emerging evidence clearly has pointed to a central role of SKP2 in regulating IFN-a/γ in the tumor microenvironment through multiple mechanisms. Therefore, Targeting SKP2 in both cancer cells and tumor microenvironment would be an excellent strategy in prostate cancer prevention and treatment.
To facilitate the efficient screening of novel selective SKP2 inhibitors, we have further developed paired prostate organoid cultures from both the hSKP2-KI and wild-type mice. While both the C1 inhibitor and the SKP2 degrader FKA selectively inhibit the growth of SKP2 overexpressing prostate organoids over normal prostate organoids at the early stage of the treatments, the SKP2 inhibitor C1 that targets SKP2/Cks1 interaction lost its activity against the growth of SKP2 overexpressing prostate organoids after 8 days of its treatments. However, FKA by degrading SKP2 protein continuously inhibits the growth of the SKP2 overexpressing prostate organoids over time. Currently, there are no effective or suitable SKP2 inhibitors for clinical trials yet. Our organoid cultures would provide a novel tool to screen and test new more potent SKP2 inhibitors for pre-clinical and clinical studies, and to understand potential mechanisms of resistance to SKP2 inhibitors with different targeting mechanisms.
In summary, the prostate-specific overexpression of hSKP2 in mice initiates spontaneous tumorigenesis in a time-dependent manner and leads to a significant remodeling of the prostatic microenvironment through inducing EMT, increasing fibroblast population and enhancing extracellular matrix accumulation. These changes are in parallel with dysregulated IFN a/γ and anti-tumor immunity signaling, enabling the evasion of immune surveillance to facilitate the process of prostate carcinogenesis. Our prostate-specific SKP2 humanized mouse model offers a valuable platform for investigating mechanisms of immune evasion and tumor microenvironment reprogramming during prostate carcinogenesis, exploring the combined effects of oncogenes and tumor suppressors, and evaluating potential agents targeting human SKP2 for prostate cancer prevention, interception and therapy.
Supplementary Material
Supplementary Files
This is a list of supplementary files associated with this preprint. Click to download.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Carrano AC, Eytan E, Hershko A, Pagano M. SKP 2 is required for ubiquitin-mediated degradation of the CDK inhibitor p 27. Nat Cell Biol 1999;1(4):193–199.10559916 10.1038/12013 · doi ↗ · pubmed ↗
- 2Lu Z, Bauzon F, Fu H, Cui J, Zhao H, Nakayama K, Skp 2 suppresses apoptosis in Rb 1-deficient tumours by limiting E 2F 1 activity. Nat Commun 2014; 5:3463.24632684 10.1038/ncomms 4463 PMC 3982150 · doi ↗ · pubmed ↗
- 3Huang H, Regan KM, Wang F, Wang D, Smith DI, van Deursen JM, Skp 2 inhibits FOXO 1 in tumor suppression through ubiquitin-mediated degradation. Proc Natl Acad Sci U S A 2005;102(5):1649–1654.15668399 10.1073/pnas.0406789102 PMC 545492 · doi ↗ · pubmed ↗
- 4Gstaiger M, Jordan R, Lim M, Catzavelos C, Mestan J, Slingerland J, Skp 2 is oncogenic and overexpressed in human cancers. Proc Natl Acad Sci U S A 2001;98(9):5043–5048.11309491 10.1073/pnas.081474898 PMC 33160 · doi ↗ · pubmed ↗
- 5ŠimečkováŠ, KahounováZ, Fedr R, Remšík J, SlabákováE, SuchánkováT, High Skp 2 expression is associated with a mesenchymal phenotype and increased tumorigenic potential of prostate cancer cells. Sci Rep. 2019;9(1):5695.30952903 10.1038/s 41598-019-42131-y PMC 6451010 · doi ↗ · pubmed ↗
- 6Wang Z, Gao D, Fukushima H, Inuzuka H, Liu P, Wan L, Skp 2: a novel potential therapeutic target for prostate cancer. Biochim Biophys Acta. 2012;1825(1):11–17.21963805 10.1016/j.bbcan.2011.09.002PMC 3242930 · doi ↗ · pubmed ↗
- 7Yang G, Ayala G, De Marzo A, Tian W, Frolov A, Wheeler TM, Elevated Skp 2 protein expression in human prostate cancer: association with loss of the cyclin-dependent kinase inhibitor p 27 and PTEN and with reduced recurrence-free survival. Clin Cancer Res 2002;8(11):3419–3426.12429629 · pubmed ↗
- 8Nguyen PL, Lin DI, Lei J, Fiorentino M, Mueller E, Weinstein MH, The impact of Skp 2 overexpression on recurrence-free survival following radical prostatectomy. Urol Oncol 2011; 29:302–308.19450994 10.1016/j.urolonc.2009.03.022PMC 5437980 · doi ↗ · pubmed ↗